
Abstract
Background
Interstitial lung disease (ILD) is a broad heterogeneous group of lung disorders that is characterized by inflammation of the lungs. Surfactant dysfunction disorders are a rare form of ILD diseases that result from mutations in surfactant protein C gene (SFTPC) with prevalence of approximately 1/1.7 million births. SFTPC patients are presented with clinical manifestations of ILD ranging from fatal respiratory failure of newborn to chronic respiratory problems in children. In the current study, we aimed to investigate the spectrum of SFTPC genetic variants as well as the correlation of the SFTPC gene mutations with ILD disease in twenty unrelated Egyptian children with diffuse lung disease and suspected surfactant dysfunction using Sanger sequencing.
Results
Sequencing of SFTPC gene revealed five variants: c.42+35G>A (IVS1+35G>A) (rs8192340) and c.43-21T>C (IVS1-21T>C) (rs13248346) in intron 1, c.436-8C>G (IVS4-8C>G) (rs2070687) in intron 4, c.413C>A p.T138N (rs4715) in exon 4, and c.557G>Ap.S186N (rs1124) in exon 5.
Conclusion
The present study confirms the association of detecting variants of SFTPC with surfactant dysfunction disorders.
Similar content being viewed by others

Background
Interstitial lung diseases (ILDs) are a heterogeneous group of lung disorders, characterized by lung inflammation and fibrosis, which result in impaired gas exchange and in progressive cases lead to respiratory failure [1]. ILDs can be caused by systemic diseases or environmental factors with a worldwide incidence of 97/100,000 cases per year [2]. SFTPC mutations are inherited in an autosomal dominant pattern, and sporadic cases with de novo mutations may occur [3]. Lung damage caused by SFTPC mutations involves a “toxic gain of function” with accumulation of the misfolded protein within type 2 pneumocytes, with subsequent injury or apoptosis consequently ending in lung fibrosis [4]. Idiopathic pulmonary fibrosis (IPF) is the most common form of ILD in adults with worldwide incidence of approximately 20/100,000 males and 13/100,000 females [5], whereas surfactant dysfunction disorders represent rare forms of ILD in both children and adults with prevalence rate of approximately 1 in 1.7 million births [6]. Clinical diagnosis of ILD requires the presence of at least three of the following criteria in the absence of known primary disorders: respiratory symptoms as nonproductive dry cough, shortness of breath or intolerance of exercise, pulmonary signs such as tachypnea, retractions crackles, failure to thrive, or finger clubbing due to hypoxemia. Extrapulmonary manifestations such as joint involvement, skin rash, and systemic hypertension were detected in some patients [7]. Family history of similar conditions, and/or history of environmental exposure to chemicals, dusts, and birds, is an important predisposing factors [8]. Diagnosis is based upon a comprehensive history, a careful physical examination, and review of laboratory data, physiologic studies, radiography, and, in some cases, pathologic tissue obtained from lung biopsy. Multidisciplinary approach is essential for sound diagnostic and management decisions involving: radiology, surgery, pathology (lung biopsy), molecular biology, and physician assessment to reach the final diagnosis. Pulmonary function testing and bronchoscopy with bronchoalveolar lavage (BAL) are important investigations recommended to rule out pulmonary vascular disease. Echocardiography is important for detection of structural cardio vascular disease or pulmonary hypertension secondary to ILD which is of important prognostic implications [9].
Systemic diseases such as autoimmune diseases, or immune deficiency disorders, should be ruled out. Genetic studies can provide the final answer in diagnosis of ILD [10]. Surfactant dysfunction disorders are caused by mutations in one of three genes: surfactant protein B (SFTPB), surfactant protein C (SFTPC), and ATP-binding cassette member A3 (ABCA3), which are essential for the function and metabolism of pulmonary surfactant. SFTPC gene mutations are inherited in an autosomal dominant pattern, while mutations of SFTPB and ABCA3 genes are inherited in an autosomal recessive pattern [11]. SFTPC gene, located on chromosome 8p21, encodes surfactant protein C (SP-C), a hydrophobic amino-acid polypeptide secreted into the alveolar space by alveolar type 2 epithelial cells to reduce surface tension [12]. ProSP-C domain plays an essential chaperone role in preventing the highly hydrophobic mature SP-C peptide from aggregating in the endoplasmic reticulum causing cell death [13]. More than 60 mutations in SFTPC gene have been identified in pediatric ILD patients, most of which are reported in patients of Caucasian or African descent with only few reports of Asian cases [14]. The second gene (SFTPB) is located on chromosome 2p11 and encodes surfactant protein B (SP-B), which is important for adsorption of secreted surfactant phospholipids to the alveolar surface [12]. The third gene is ATP-binding cassette transporter A3 (ABCA3) is expressed in type 2 alveolar epithelial cell lamellar bodies and plays an important role in pulmonary surfactant synthesis and transport [15]. There are nonspecific curative therapies for ILD, and only a few therapies might slow disease progression; however, management decisions would vary depending on ILD subtype [16]. Lung transplantation is possible for only urgent cases of idiopathic pulmonary fibrosis (IPF) patients [17]. The present study explores the association of SFTPC genetic variations with suspected surfactant dysfunction diseases in a cohort of Egyptian children.
Methods
Patients
Twenty unrelated ILD patients were enrolled in the present study after obtaining an informed consent from their guardians following the guidelines of the Institution Review Board. The included patients were referred from the outpatient allergy and pulmonology clinic, Abo El Reesh Hospital to the Clinical Genetics Clinic, Center of Excellence of Human Genetics. All patients were subjected to full history taking, family pedigree analysis, and thorough examination of all body systems with special emphasis on chest examination.
Clinical criteria
The 20 studied probands were included based on presenting with breathing difficulties including the following: shortness of breath, coughing typically nonproductive, decreased exercise tolerance, fatigue, and/or weight loss. The excluded patients were those with an underlying cause of interstitial lung diseases such as immunodeficiency, collagen vascular disorders, environmental exposure, or gastroesophageal reflux disease (GERD).
Molecular analysis
Genomic DNA was extracted from all 20 patients’ peripheral blood using Thermo Scientific GeneJET Genomic DNA Purification Kit (USA) according to manufacturer’s instructions. All the coding region and exon-intron boundaries were amplified with specific primers of SFTPC gene. The sequences of the designed primers are available on request. PCR cycling conditions were generated in (Perkin-Elmer Cycler, USA) as follows: 94 °C for 10 min followed by 35 cycles of 94 °C for 1 min, 59 °C for 1 min, and 72 °C for 1 min, followed by final cycles are extension at 72 °C for 10 min. The PCR products were purified using the ExoSAP Cleanup kit (Fermentas, Germany) and sequenced in both directions using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) and analyzed on the ABI Prism 3500 Genetic Analyzer (Applied Biosystems, USA). Sequencing results were then analyzed on the NCBI website (http://blast.ncbi.nlm.nih.gov/Blast.cgi) and compared with cDNA sequence of FTPC gene (NM_003018.4).
Results
Clinical results
The twenty studied patients were descending from unrelated 20 pedigrees; positive consanguinity was detected in 11 out of the twenty included patients’ families.
Our studied cohort included 11 males and 9 females whose ages at examination ranged from 1 and 6/12 to 12 years. All patients were presenting with clinical criteria suggestive of ILD including cough and progressive dyspnea. Clubbing of fingers and toes was detected in elder patients. Short stature and failure to thrive were detected in 6/20 (30%) patients. Clinical data of all patients is summarized in Table 1.
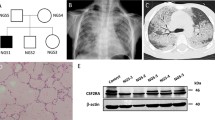
Plain chest radiographic examinations showed patchy infiltrates in the lungs of all the included patients (Fig. 1). High-resolution computed tomography (HRCT) revealed ground glass attenuation, honeycombing, and cyst formations. Ground glass attenuation opacities in CT chest were evident in our patients (Figs. 2 and 3). The echocardiographic evaluation detected pulmonary hypertension in 9 of the included patients and congenital heart diseases in the other 2 patients in the form of patent ductus arteriosus (PDA) and patent foramen oval (PFO).

Plain chest radiographic examination showing patchy infiltrates in the lungs

Bilateral lower lobar, to less extent middle lobar diffuse ground glass attenuation, reticulonodular densities, and peribronchial thickening (HRCT patient number 4)

Ground glass appearance, peribronchial thickening, and cyst formation bilateral (HRCT patient number 5)
Results of molecular analysis
Sanger sequencing of SFTPC gene of the twenty Egyptian ILD patients revealed five different variants in 17/20 unrelated patients: three intronic and two exonic variants. Three intronic sequence variants included the following: c.42+35G>A (IVS1+35G>A) (rs8192340) in one patient and c.43-21T>C (IVS1-21T>C) (rs13248346) in six patients, both variants were located in intron 1, while c.436-8C>G (IVS4-8C>G) (rs2070687) in intron 4 was detected in one patient. The two exonic missense variations included the following: c.413C>A (rs4715) in exon 4 disclosed from five patients resulting in the exchange of threonine by asparagine at codon 138 (p.T138N) and exon 5, c.557G>A (rs1124) gene variation, were detected in four patients resulting in substitution of serine with asparagine at codon 186 (p.S186N) (Figs. 4 and 5). No more variants were detected within the remaining three patients within SFTPC gene (Table 2).

Schematic representation of localization of five reported variants in SFTPC gene: c.42+35G>A and c.43-21T>C in intron 1, c.436-8C>G in intron 4, c.413C>A in exon 4, and c.557G>A in exon 5

Sequence chromatograms showing five reported variants in SFTPC gene. A Normal and variant of (rs8192340) c.42+35G>A in intron 1, B normal and variant of (rs13248346) c.43-21T>C in intron 1, C normal and variant of (rs2070687) c.436-8C>G in intron 4, D normal and variant of (rs4715) c.413C>A in exon 4, and E normal and variant of (rs1124) c.557G>A in exon 5
Discussion
Interstitial lung diseases (ILDs) are a heterogeneous group of diseases characterized by widespread fibrotic and inflammatory abnormalities of the lung. Many forms of ILD are extremely rare, while other forms such as idiopathic pulmonary fibrosis (IPF) and sarcoidosis are seen commonly in general pulmonary practice. ILD refers to a heterogeneous collection of more than 200 distinct lung disorders that tend to be grouped together because they share clinical, radiographic, and pathologic features. These diseases have a broad spectrum of clinical presentations and manifestations ranging from lethal neonatal respiratory distress syndrome to adult chronic interstitial lung disease (ILD). In general, most interstitial lung diseases are characterized by four manifestations: (1) respiratory symptoms such as shortness of breath and cough, (2) specific chest radiographic abnormalities, (3) typical changes on pulmonary function tests in which the lung volume is decreased, and (4) characteristic microscopic patterns of inflammation and fibrosis [18,19,20].
Interstitial lung disease (ILD) in infants and children is associated with affected growth and high morbidity and mortality. All patients were recurrently admitted to hospital due to recurrent pneumonia, and they continued to have cough and tachypnea, and some of them exhibited failure to thrive later on [21].
Radiological investigations (chest X-ray) are the first approach as it is frequently abnormally diffused. But it has limited and low diagnostic specificity. Chest x-ray was performed for all studied 20 cases and showed patchy lung infiltrates. High-resolution computed tomography (HRCT), as a better diagnostic imaging technique for ILD children, helps avoid the need for surgical lung biopsy and is also useful in monitoring the therapeutic response. The most commonly observed findings in ILD by this technique are ground glass attenuation, honeycombing, and cyst formations. Ground glass attenuation opacities in CT chest were evident in all our patients and were our first clue for clinical diagnosis of ILD [22].
Echocardiography performed to the probands diagnosed 9 cases with pulmonary hypertension and 2 cases with congenital heart disease (patent ductus arteriosus (PDA) and patent foramen oval (PFO)); these findings were similar to a study, reporting a newly born female infant with neonatal pneumonia and PFO (1.9 mm). For some types of pediatric ILDs and few forms in adult ILDs, genetic causes have been identified and precise. Molecular diagnosis was used instead of the need for lung biopsies allowing definite diagnosis accessible to clinicians. Understanding the mechanisms of the idiopathic forms of interstitial lung disease is currently emerging. Studies of cells culture revealed a number of molecules and molecular pathways (such as transforming growth factor beta) that promote fibrosis [14, 23].
Previous studies reported genetic correlation of idiopathic pulmonary fibrosis (IPF) alterations in specific genes such as surfactant protein genes and telomerase. Further evidence for the role of genetics in interstitial lung disease comes from studies in patients with other disorders as sarcoidosis and Hermansky–Pudlak syndrome, where mutations in specific genes are associated with a higher incidence of lung fibrosis [24].
Egyptian study, conducted on 568 cases (191 males and 377 females) with mean age 44 ± 12 years from Upper Egypt, noted that IPF is the most common lung disease due to domestic air pollution, indoor exposures, and environmental factors [25].
Monoallelic mutations of the surfactant protein C gene (SFTPC) were associated with (ILD) in children and adults. When the SP-C gene is mutated, the precursor of surfactant protein C (proSP-C) is misfolded and accumulates within the ER and Golgi apparatus of AEC2s, leading to cellular injury and apoptosis [14].
In the present study, sequencing analysis of SFTPC gene within 20 ILD patients revealed five variants: c.42+35G>A (rs8192340), c.43-21T>C (rs13248346), c.436-26C>G (rs2070687), c.413C>A (rs4715), and c.557G>A (rs1124). A similar study was carried out on 760 Caucasian of Danish descents with different lung disease phenotypes as asthma, chronic obstructive pulmonary disease (COPD), and interstitial lung disease; they identified 18 variants including our five detected variants [26].
In patients with exonic mutations, the two previously reported SFPTC mutations in humans were described as acting in dominant fashion, with abrogation of SP-C synthesis (dominant negative effect). However, no deviation from the reference sequence was observed among patients harboring the variants predicted as benign after thorough SFTPC analysis, suggesting that either a cryptic SFTPC mutation segregated in this family, or that the SP-C deficiency was caused by a mutation at another genetic locus or by shared environmental exposures. Also, robust linkage disequilibrium was detected between the coding SNPs, T138N, and S186N by where the occurrences of the predictable haplotypes (for the allele frequencies of both SNPs) did not change considerably between patient and control groups. Remarkably, diverse histopathological types of pulmonary fibrosis were discovered in members of the same kindred harboring the same SFTPC mutation, and these diverse types were interpreted as possibly signifying pleiotropic expressions of the same genetic defect. SFTPC-associated familial forms of pulmonary fibrosis are characterized by reduced penetrance suggesting that further endogenous or exogenous elements might back the marked variety of pulmonary fibrosis predisposed by SFTPC mutations [13, 14, 27, 28].
In our study, single-nucleotide variants c.42+35G>A (rs8192340) and IVS4-8C>G (rs2070687), each was detected in two different patients (4 & 11); these variants were reported previously in 2 unrelated adult Dutch patients with familial pulmonary fibrosis though their mean ages at diagnosis were 54 years [29].
IVS4-8C>G (rs2070687) variant was reported in two American infants in two different studies; this variant was associated with persistent pulmonary hypertension (PPHN) and respiratory distress syndrome (RDS) symptoms [30, 31].
Sequencing analysis of (SFTPC) gene revealed two common missense variants p.T138N (rs4715) in exon 4 in five patients and S186N (rs1124) in exon 5 in four patients. p.T138N was predicted to be possibly damaging, affecting the domain of precursor SP-C (proSP-C) using in silico analysis, while the S186N variant was predicted to be polymorphism with no pathogenic effect [32].
The two common variants p.T138N and p.S186N in SFTPC gene were reported in two different studies, one in adult German (IPF) patients with age ranging from 24 to 61 years and the other in familial pulmonary fibrosis cases within population of Reunion Island, represented with unexplained respiratory distress (URD) [27, 28].
Moreover, the two variants p.T138N and p.S186N were described as minor alleles in two unrelated Finnish infants suffering respiratory distress syndrome (RDS) whose ages were less than 34 weeks with frequencies 0.20 and 0.22, respectively. Meanwhile, another study included 100 American patients assigned with familial pulmonary fibrosis, investigating the same two variants in frequencies of 0.255 and 0.318, respectively [33, 34].
Heterozygous SFTPC mutations concomitant, as well as heterozygous mutations in ATP-binding cassette transporter A3 (ABCA3) in infants with interstitial lung diseases (ILD), might likely led to development of clinical ILD [35].
Advances in understanding genetic factors contributing to ILD could outline some of the emerging roles of epigenetic modifications and give a vision about the progressed directions towards future prospects of genetically targeted therapies [18, 24].
Whole-exome sequencing (WES) has become an advanced approach to investigate rare alleles with direct functional consequences on protein products, affecting their pathways. WES was used to screen mutations concerned with surfactant proteins A and C (SFTPA2 and SFTPC) genes; these two genes were mainly related to cell adhesion and immune response, which might partially explain changes of gene expression involved in immune-related pathways in ILD [36].
Molecular studies were performed using the next-generation sequencing of surfactant dysfunction genes identified three mutations in surfactant protein-C gene (SFTPC) in 6 Chinese children with ILD symptoms, whose ages of onset ranged from 7 days to 15 months: I73Tin 4/6, D105G in 1/6, and Y113H in 1/6 patients [37].
Our study highlights five variants: c.42+35G>A (IVS1+35G>A) (rs8192340) and c.43-21T>C (IVS1-21T>C) (rs13248346) in intron 1, c.436-8C>G (IVS4-8C>G) (rs2070687) in intron 4, c.413C>A p.T138N (rs4715) in exon 4, and c.557G>Ap.S186N (rs1124) in exon 5 within SFTPC gene that were all associated with interstitial lung disease (ILD) in Egyptian children.
Conclusions
ILD comprises a heterogeneous group of diffuse parenchymal lung processes with overlapping clinical and histopathologic features. Progress has been made in identifying genes and pathways critical for ILD; the molecular and genetic causes of most lung malformations affecting lung function remain to be elucidated. Although the current study prides a diagnostic pilot seed, further studies should be carried out on a larger scale of Egyptian cases with symptoms of interstitial lung disease (ILD) to investigate all surfactant dysfunction causative genes (SFTPA1, SFTPA2, SFTPB, SFTPD, ABCA3, TERT, TERC, TINF2, PARN, NAF1, and MUC5B) by performing NGS-customized panel or whole-exome sequencing techniques. Advanced validation studies with larger statistical power are required to verify de novo findings and identify the underlying functional pathways. This will improve knowledge on the pathogenesis of associated surfactant diffuse lung disease in children and provide ILD families with a precise diagnosis as well as better genetic counseling.
Availability of data and materials
The data sets generated and/or analyzed during the current study are not publicly available due to patient’s privacy but are available from the corresponding author upon request.
Change history
21 September 2022
A Correction to this paper has been published: https://doi.org/10.1186/s43141-022-00420-6
Abbreviations
- ABCA3 :
-
ATP-binding cassette transporter A3
- BAL:
-
Bronchoalveolar lavage
- COPD:
-
Chronic obstructive pulmonary disease
- FTT:
-
Failure to thrive
- GERD:
-
Gastroesophageal reflux disease
- HRCT:
-
High-resolution computed tomography
- ILD:
-
Interstitial lung disease
- IPF:
-
Idiopathic pulmonary fibrosis
- PDA:
-
Patent ductus arteriosus
- PFO:
-
Patent foramen ovale
- PH:
-
Pulmonary hypertension
- PPHN:
-
Persistent pulmonary hypertension
- RDS:
-
Respiratory distress syndrome
- SP-B:
-
Surfactant protein B
- SP-C:
-
Surfactant protein C
- SFTPB :
-
Surfactant protein B gene
- SFTPC :
-
Surfactant protein C gene
References
Borie R, Guen PL, Ghanem M et al (2019) The genetics of interstitial lung diseases. Eur Respir J 28:190053
Duchemann B, Maesano IA, Naurois CJ et al (2017) Prevalence and incidence of interstitial lung diseases in a multi-ethnic county of Greater, Paris. Eur Respir J 50:1602419
Kaleel M, Schramm C, Pascal M et al (2015) Serial lung magnetic resonance imaging to monitor disease progression in a child with a diffuse alveolarhemorrhagesyndrome. J Clin Med Res 7(4):267–269
Mulugeta S, Nguyen V, Russa SJ, Muniswamy M et al (2005) A surfactant protein C precursor protein BRICHOS domain mutation causes endoplasmic reticulum stress, proteasome dysfunction , and caspase 3 activation. J Respir Cell Mol Biol 32(6):521–530
Hutchinson J, Fogarty A, Hubbard R et al (2015) Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J 46:795–806
Garmany TH, Wambach JA, Heins HB et al (2008) Population and disease-based prevalence of the common mutations associated with surfactant deficiency. Pediatr Res 63:645–649
Cazzato S, Palmo E, Ragazzo V et al (2013) Interstitial lung disease in children. Early Hum Dev 89(3):S39–S43
Ishak SR, Hassan AM, Kamel TB (2021) Environmental hazards and demographic and clinical data of childhood interstitial lung diseases in a tertiary institute in Egypt. Egypt J Bronchol 15:2
Fan LL, Kozinetz CA (1997) Factors influencing survival in children with chronic interstitial lung disease. Am J Respir Crit Care Med 156(3 pt 1):939–942
Dinwidie R, Sharief N, Crawford O (2002) Idiopathic interstitial pneumonitis in children: a national survey in the United Kingdom and Ireland. Pediatr Pulmonol 34(1):23–29
Gower WA, Nogee LM (2011) Surfactant dysfunction. Pediatr Res Rev 12(4):223–229
Guillot L, Epaud R, Thouvenin G et al (2009) New surfactant protein C genemutations associated with diffuse lung disease. J Med Genet 46(7):490–494
Wert SE, Whitsett JA, Nogee LM (2009) Genetic disorders of surfactant dysfunction. Pediatr Dev Pathol 12(4):253–274
Hong D, Dai D, Liu J et al (2019) Clinical and genetic spectrum of interstitial lung disease in Chinese children associated with surfactant protein C mutations. Ital J Pediatr 45:117
Zhou W, Zhuang Y, Sun J et al (2017) Variants of the ABCA3 gene mightcontribute to susceptibility to interstitial lung diseases in the Chinese population. Sci Rep 7:4097
King TE, Bradford WZ, Castro-Bernardini S et al (2014) A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 370:2083–2092
Richeldi L, du Bois RM, Raghu G et al (2014) Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 370:2071–2082
American Thoracic Society, European Respiratory Society. American Thoracic Society/ European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias (2002) This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors. Am J Respir Crit Care Med 165:277–304
Willander H, Askarieh G, Landreh M et al (2012) High-resolution structure of a BRICHOS domain and its implications for anti-amyloidchaperone activity on lung surfactant protein C. Proc Natl Acad Sci 109:2325–2329
Thillai M, David MR, Keith MC (2017) Clinical Handbook of Interstitial Lung Disease, 1st edn. United States of America: CRC Press
Nogee LM, Dunbar AE, Wert SE et al (2001) A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med. https://doi.org/10.1056/NEJM20010222.344.0805
Kurland G, Deterding RR, Hagood JS et al (2013) An official American Thoracic Society clinical practice guideline: classification, evaluation and management of childhood Interstitial Lung Disease. Am J Respir Crit Care Med. https://doi.org/10.1164/rccm.201305-0923ST
Nogee LM (2002) Abnormal expression of surfactant protein C and lung disease. Am J Respir Cell Mol Biol 26:641–644
Steele MP, Brown KK (2007) Genetic predisposition to respiratory diseases: infiltrative lung diseases. Respiration 74:601–608
Rashad MA, Ibrahim AK (2015) Idiopathic pulmonary fibrosis (IPF) in Upper Egypt, a single center study, Egypt. J Chest Dis Tuberc. https://doi.org/10.1016/j.ejcdt.2015.05.007
Baekvad-Hansen M, Børge G, Nordestgaard BG et al (2010) Lung function and obstructive lung disease. Respir Med 104:418–425
Markart P, Ruppert C, Wygrecka M et al (2006) Surfactant protein C mutations in sporadic forms of idiopathic interstitial pneumonias. ERJ Express. https://doi.org/10.1183/09031936.00034406
Tredano M, Griese M, Brasch F, Couderc R, Bahuau M et al (2004) Mutation of SFTPC in infantile pulmonary alveolar proteinosis with or without fibrosing lung disease. Am J Med Genet 126:18–26
Van M, Oosterhout V, Barlo NP et al (2010) Surfactant protein C mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a Dutch cohort. Am J Respir Crit Care Med 182:1419–1425
Byers MH, Dagle MJ, Klein MJ et al (2012) Variations in CRHR1 are associated with persistent pulmonary hypertension of the newborn. Pediatr Res 71(2):162–167
Ryckman KK, Dagle HM, Kelsey K et al (2012) Genetic Associations of surfactant protein D and angiotensin- converting enzyme with lung disease in preterm neonates. J Perinatol 32(5):349–355
Chun-mei Z, Ling C, Rong-yan H et al (2013) Pulmonary surfactant protein gene mutation associated with pediatric interstitial lung disease: a case study and the review of related literature. Chin J Pediatr 51(2):84–89
Wambach JA, Yang P, Wegner DJ et al (2010) Surfactant protein-C promoter variants associated with neonatal respiratory distress syndrome reduce transcription. Pediatr Res 68:216–220
Maitra M, Dey M, Yuan W, Peter W et al (2013) Lung fibrosis-associated surfactant protein A1 and C variants induce latent transforming growth factor _1 secretion in lung epithelial cells. J Biol Chem 288(38):27159–27171
Bullard JE, Nogee LM (2007) Heterozygosity for ABCA3 mutations modifies the severity of lung disease associated with a surfactant protein C gene (SFTPC) mutation. Pediatr Res 62:176–179
Fang C, Huang H, Feng Y et al (2021) Whole-exome sequencing identifiessusceptibility genes and pathwaysfor idiopathic pulmonary fibrosisin the Chinese population. Sci Rep 11:1443
Hong D, Dai D, Liu J et al (2020) Clinical and genetic spectrum of interstitial lung disease in Chinese children associated with surfactant protein C mutations. Ital J Pediatr 45:117
Acknowledgements
Not applicable
Funding
Not applicable
Author information
Authors and Affiliations
Contributions
AKA, EAA, and MME performed clinical selection, evaluation, and documentation of clinical criteria of the patients in the manuscript. MOR, SAES, and GEK interpreted clinical criteria and revised the manuscript. MMR performed the molecular and software analysis as well as wrote the manuscript. KSA analyzed and interpreted the results and revised the manuscript. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The research protocol was approved by the National Research Centre (NRC) Institutional Review Board. Patients were referred from the outpatient allergy and pulmonology clinic, Abo El Reesh Hospital, Cairo University to the Clinical Genetics Clinic, Center of Excellence of Human Genetics, NRC, Cairo, Egypt. Sharing was voluntary; an informed written agreement was obtained from each participant before enrolment into the study. Data were anonymous and coded to assure confidentiality of participants. Also, written informed consent was obtained from parents, legal guardian of participants under 16 years old. Committee’s reference number is not applicable.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: the authors identified an error in the Authors’ contributions section for Engy A. Ashaat (EAA).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abdel Megeid, A.K., Refeat, M.M., Ashaat, E.A. et al. Correlating SFTPC gene variants to interstitial lung disease in Egyptian children. J Genet Eng Biotechnol 20, 117 (2022). https://doi.org/10.1186/s43141-022-00399-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43141-022-00399-0