
Abstract
Background
DNA barcoding is a powerful method for phylogenetic mapping and species identification. However, recent research has come to a consistent conclusion about the universality of DNA barcoding. We used matK and rbcL markers to test the universality of twelve accessions from different locations belonging to two Selinum species, Selinum tenuifolium Wall. C. B. Clarke and Selinum vaginatum C. B. Clarke, keeping in mind their ability to identify species and establish phylogenetic relationships within and between the accessions.
Results
The success rates of PCR amplification using matK and rbcL were 75.26% ± 3.65% and 57.24% ± 4.42%, and the rate of DNA sequencing was 63.84% ± 4.32% and 50.82% ± 4.36%, respectively, suggesting that success rates of species identification of the two fragments were higher than 41.00% (matK, 41.50% ± 2.81%; rbcL, 42.88% ± 2.59%), proving that these fragments might be used to identify species. The best evolutionary tree with good supporting values was produced utilizing combinations of matK + rbcL markers when phylogenetic relationships were built with random fragment combinations. The twelve accessions of Selinum collected from different locations and their molecular sequences of matK and rbcL markers were blasted with other genera of Apiaceae family, and it was found that Selinum is most closely related to Angelica species of Apiaceae family.
Conclusion
The present study has grouped twelve accessions of Selinum species using molecular markers into phylogenies, which is first-of-its-kind report that established interrelationships within different species of Apiaceae with respect to Selinum.
Graphical abstract

Similar content being viewed by others


Background
Since thousands of years, medicinal plants have been used in health-care systems [1, 2]. The correct identification of these plants is required for their safe use. Medicinal plants were traditionally classified by experienced experts based on morphological characteristics. However, it may take an expert many years to accurately identify these plants, and only a few taxonomists have been able to do so for around 1000 species so far [3]. Furthermore, the number of trained experts has been decreasing as the workload on identification has increased. Simultaneously, the number of adulterants and substitutes on the market is increasing, resulting in variation in the quality of medicinal plants [4, 5]. Thus, identifying medicinal plants is a significant challenge, posing safety risks to both the traditional medicine industry and consumers. As a result, a quick and accurate method for authenticating these plant species is required. DNA barcoding is a novel method for species identification that has the capacity to overcome the aforementioned challenges [3].. This method is based on sequence differences within short, standardized DNA regions, with the primary goal of both authenticating and discovering new species [3, 6, 7]. DNA barcoding is an efficient and low-cost technique that could boost the work of specialists while also making species validation accessible to nonspecialists [8,9,10,11]. Furthermore, it is independent of both morphological characteristics and expert experiences, allowing for faster, more subjective, and accurate results. As a result, DNA barcoding has quickly become the focus of research, and it has been recognized as a powerful tool for species identification and the construction of phylogenetic trees [12, 13].
The Apiaceae family contains approximately 250–440 genera and a range of 3300–3700 species that are widely distributed throughout the temperate zones of both hemispheres. Selinum belongs to Apiaceae is perennial, aromatic, and medicinal plant ranging from 1 to 2 m in length [14]. Selinum species are mainly found in Western Himalayas and Himalayan foothills of India, West Pakistan, Bhutan, China, and Nepal. Some of its species have also been reported from Europe, specifically in the UK, Poland, Bosnia, and Croatia. Ecological and floral diversity type studies so far do not present the molecular characterization of the Selinum spp. [15, 16]. Molecular characterization may further provide database for intensive pharmacological studies in Selinum.
It has proven difficult to classify species within the Selinum genus because there are no clear morphological synapomorphies and the genus is plagued by synonymies, in which a species is referred to by a different name somewhere else [17]. Several other Apiaceae genera, such as Angelica L., Ferula L., and Coriandrum sativum L., are also treated separately; however, no phylogenetic studies are available in Selinum.
Two species of Selinum, viz S. tenuifolium Wall ex C. B. Clarke (Milk Parsley) and S. vaginatum C. B. Clarke (Bhutkeshi), have been identified on the basis of morphology in India. The National Center for Bioinformatics database and UniProt database reveal that studies have only been done on specified regions of the gene or proteins of the study with pharmaceutical or cosmetic motives. However, they do not provide molecular characteristics of Selinum spp. [18]. Currently, rbcL genes are widely used for phylogenetic analysis within angiosperm families and subclasses, as well as among seed plant groups. However, matK evolves 3 times faster than rbcL in angiosperms, potentially providing more phylogenetic information and improved phylogenetic resolution among the taxa studied [19, 20]. The present study therefore aims to assess the molecular characteristics of Selinum spp. using matK and rbcL plastid genes in order to derive a phylogenetic relationship within the sp. of Selinum as well as with other members of Apiaceae.
Methods
Sample collection
The plants were sampled from twelve accessions of Selinum collected from different locations representing two states in the Northern Himalayan region of India, namely Himachal Pradesh (HP) and Uttarakhand (UK). Seven S. vaginatum C. B. Clarke accessions were collected from various locations, viz SV1 — S. vaginatum, Chipla kedar, 2691 m (UK); SV2 — S. vaginatum, Rohtang valley, 2137 m (HP); SV3 — S. vaginatum, Dhartula, 2378 m (UK); SV4 — S. vaginatum, Cheena Peak, 1937 m (UK); SV5 — S. vaginatum, Dhartula, 1819 m (UK); SV6 — S. vaginatum, Chipla Kedar, 2236 m (UK); and SV7 — S. vaginatum, Gulaba 2781 m (HP), and five accessions of S. tenuifolium Wall ex C. B. Clarke were collected from different locations (ST1 — S. tenuifolium, Kothi, 2158 m (HP); ST2 — S. tenuifolium, Rohtang valley, 2350 m (HP); ST3 — S. tenuifolium, Chopta trek, 2150 m (UK), ST4 — S. tenuifolium, Chopta trek, 2593 m (UK), ST5 — S. tenuifolium, Chopta trek, 3178 m (UK)) and stored at −4 °C for molecular studies.
For two target genes, matK and rbcL, species disparity was accomplished through BLASTn analysis and phylogenetic inference based on maximum likelihood and neighbor-joining method. The disparity index was used to assess the dependability of phylogenetic tree regenerations. The maximum composite likelihood with Kimura 2-parameter model was used to estimate evolutionary divergence between and within all accessions.
Laboratory reagents, plasticware, and glassware
SIGMA (Sigma-Aldrich, Inc., 3050 Spruce Street, St. Louis, MO, USA) and Gyan Scientific analytical grade reagents and chemicals were used in this study (Lucknow, India). Borosil glassware and Axygen plasticware were used.
Sterilization of plasticware and glassware
The glassware and plasticwares used for our experiment purpose were washed, cleaned, and sterilized as per the standard protocol recommended for molecular biology researches. All the glassware and plasticware were autoclaved before use, and UV (ultraviolet) irradiation was used as a standard laboratory practice to decontaminate the apparatus. Carryover of DNA from such a previous amplification of the same target almost always results in false-positive amplification. To prevent this, PCR reactions were set up in a separate room than that used for post-PCR manipulations. Autoclaved deionized water (Millipore ZLXS50034, Millipore Bangalore, India) was used for conducting all experiments.
Genomic DNA extraction
Five S. tenuifolium accessions and seven S. vaginatum accessions were collected, and their genomic DNA was isolated from young leaves and used as a template for a PCR reaction using a DNA isolation kit (QIAGEN, DNeasy® Plant Handbook). With the respective universal primers, the target DNA regions, namely matK and rbcL, were amplified.
Bioinformatics tools
The software used for analyzing the chromatograms of amplicons generated after sequencing by ABI 3130 Genetic Analyzer (Applied Biosystems Inc., USA) was as follows:
-
a)
BioEdit program version 3 (Naim & Mahboob. 2020) [21]
-
b)
Tamura 3-parameter model (Olmstead & Palmer 1994) [22]
-
c)
MEGA version 6.06 (Hartl & Frost 1999) [23]
Barcoding gene for the study
To evaluate intraspecific and interspecific variations among the accessions of genus Selinum, two genes of DNA barcodes (rbcL, matK) were selected to identify diversity among selected species.
PCR amplification with matK and rbcL primers
A reaction mixture containing 50 μl of Taq PCR Master Mix to conduct PCR, 22 μl of distilled water, 1 μl of forward primer (10 μM), 1 μl of reverse primer (10 μM), and 1 μl of DNA template (50–80 ng/l) was used to conduct PCR. The PCR conditions are mentioned in Table 1, and primer details are given in Table 2.
The PCR products were tested on a 1.5% agarose gel with ethidium bromide in 1X TAE buffer (pH 8.0) to ensure that they were correct. Molecular Imager® GelDocTMXR software was used to examine and archive the gel. With Quantity One software, bands were scored and evaluated (Bio-Rad). By comparing the size of the products to 500 bp DNA Ladder H3 RTU, the size of the products was determined (GeneDirex, cat no. DM003-R500) (Figs. 1 and 2). The nucleotide sequence was converted into an amino acid sequence after the PCR products were verified by electrophoresis, purified, and sequenced using the following comprehensive sequencing procedures (Table 3). The nucleotide and amino acid sequences were aligned using the system software aligner tool to produce a phylogenetic tree based on two statistical data analyses, bootstrapping and pairwise distance, from both nucleotide and amino acid sequences. Table 4 shows the sequencing and features in greater detail.

Agarose gel showing amplification of rbcL gene

Agarose gel showing amplification of matK gene
Results
Evolutionary and phylogenetic analysis
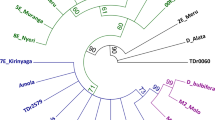
After DNA sequencing, the twelve accessions of Selinum collected from different locations and their molecular sequences of matK and rbcL markers were evaluated using BLAST method. As a statistical method and a phylogeny test, aligned sequences were used to create phylogenetic trees using the maximum composite likelihood model and bootstrap resampling, respectively. Gene sequences from the twelve Selinum accessions were used to create the most likely phylogenetic trees. The numbers at branch nodes are bootstrap values, which show the percentage of bootstrap iterations that support the tree at that specific point of divergence for each phylogenetic tree node. The more the topology of the phylogenetic tree is supported, the higher the bootstrap value. In the phylogenetic tree, species that are adjacent to each other are closely linked. ST5, SV4, ST4, and SV5 have been demonstrated to be closely related (cluster 1) and have a moderate bootstrap support value of 49, whereas ST2, ST1, ST3, SV2, SV6, and SV7 have been grouped together (cluster 2) and have a bootstrap value of 41, showing that this grouping is poorly supported. ST5 is a distant common ancestor of all the accessions. ST3, SV3, SV6, ST4, and SV5 have also been grouped together (cluster 3), while SV7, SV1, SV4, ST1, ST2, and SV2 have been grouped together (cluster 4) based on rbcL gene sequences, and both groupings are related to SV1 and SV3, which are clustered separately. Cluster 3 has a moderate bootstrap support rating of 43, whereas cluster 4 has a high bootstrap value of 53, indicating that it is strongly supported. The evolutionary trees of matK and rbcL are surprisingly similar. ST5, SV4, ST4, and SV5 have been grouped together based on matK genes, and all four accessions form a monophyletic group (cluster 1). The phylogenetic tree built using the rbcL genes shows the similar pattern (cluster 4). ST2, ST1, ST3, SV2, SV6, and SV7 have all been demonstrated to be closely related using matK genes (cluster 2). Such species’ close association can also be seen in phylogenetic trees based on the rbcL genes (cluster 4). ST2 has been shown to be closely associated with ST1, ST3, SV2, SV6, and SV7 (cluster 2) based on matK genes, whereas SV7 has been shown to be closely related to SV1, SV4, ST1, ST2, and SV2 (cluster 4) based on rbcL genes, as shown in Fig. 3.

Comparative analysis of 12 different accessions of Selinum from matK and rbcL markers
Assessment of genetic diversity in Selinum using matK and rbcL markers
Nucleotide variation
The length of the matK gene sequence ranged from 740 to 855 base pairs (bp), while the rbcL gene ranged from 582 to 638 bp. DNA base composition represented as G + C content, where G + C percentage in all species is 40% on average. AT (adenine-thymine) was dominant over GC (guanine-cytosine) in the composition of nucleotides from both genes. In rbcL, GC content was 56% and AT was 44%, while in matK, GC content was 65% and AT was 35%, respectively (Fig. 4 and Table 5).

GC content distribution of matK (upper panel) and rbcL (lower panel) in sequences gene
Phylogenetic relationship between the Selinum species
Based on partial rbcL gene sequences and matK gene sequences, the tree construction findings were midpoint rooted. Molecular phylogenetic trees for the rbcL and matK gene constructed using the maximum likelihood (ML) methods showed approximately identical clustering (Fig. 5). The trees showed that two main divisions of the chosen accessions from different locations of Selinum group were clustered together. Four clusters emerged from a cluster analysis of data gathered from the matK gene in twelve Selinum accessions from various regions. Angelica sylvestris was in the first cluster (ST1). The second cluster included Peucedanum ostruthium (ST4), Peucedanum ostruthium (SV5), and Angelica sylvestris (ST2). The third cluster included Seseli libanotis (ST3) and Peucedanum ostruthium (SV4), while the fourth cluster included Angelica sylvestris (ST5), Seseli libanotis (SV6), Seseli libanotis (SV7), Angelica sylvestris (SV2), Angelica sylvestris (SV3), and Peucedanum ostruthium (SV1) as shown in Fig. 5, and cluster was given in Table 6.

1000 bootstrap replications of phylogenetic tree of partial matK gene sequences
Clustering analysis from the rbcL gene, using S. vaginatum as the out-group and rbcL phylogenetic trees, found two primary clades and one out-group clade. Seseli libanotis is the first clade (SV1). Clade two consist of four accessions: Peucedanum palustre (ST5), Peucedanum palustre (ST1), Peucedanum palustre (SV2), and Peucedanum palustre (ST2), while clade 3 consists of seven accessions, Peucedanum palustre (ST3), Peucedanum palustre (SV7), Peucedanum palustre (SV6), Peucedanum palustre (SV5), Peucedanum palustre (ST4), Peucedanum palustre (SV4), and Peucedanum palustre (SV3) as shown in Fig. 6 and Table 7.

Using 1000 bootstrap replications, phylogenetic tree of partial rbcL gene sequences
Inter- and intraspecific relationship between matK and rbcL
Based on phylogenetic analysis, twelve accessions, including ST1, ST2, ST3, ST4, ST5, SV1, SV2, SV3, SV4, SV5, SV6, and SV7 from different geographical locations of India, were slightly different from all other accessions and were grouped together (Fig. 3). The samples were also divided into two clades using the maximum likelihood method. Clade 1 was made up of a monophyletic clade that included all of the samples under consideration. The first subgroup was further split into two clades. Clade 1 nested all the matK samples under it, while only a single rbcL sample was under clade 2. Similarly, group 2 also has two main clusters, both having largely rbcL samples.
The intraspecific and interspecific analysis of twelve accessions from different locations of Selinum infer that the intraspecific variation between the accessions is very less and interspecific variation as exhibited by matK and rbcL marker showed variations of different degrees among the species (Fig. 7). matK markers showed maximum interspecific variation (97%) and rbcL markers only 3% (Fig. 8). Intraspecific variation was also maximum in matK (61%) marker fallowed by rbcL (39%) (Fig. 9).

Interspecific and intraspecific relationships between matK and rbcL gene of twelve accessions of S. tenuifolium and S. vaginatum collected from different locations

Interspecific relationships between matK and rbcL gene of twelve accessions of S. tenuifolium and S. vaginatum collected from different locations

Interspecific relationships between matK and rbcL and between matK and rbcL gene of twelve accessions of S. tenuifolium and S. vaginatum collected from different locations
Discussion
The current study found that the DNA identification approach appropriately differentiated all samples up to species level, and that matK and rbcL are reliable identification markers [21]. It is an accurate method to amplify and sequence, and plastid DNA has played a significant role in building evolutionary relationships and defining limits of species in plants. It is also mostly uniparentally inherited and appears to have little to no recombination, resulting in a small size, structure, and gene order [22]. It is critical to understand the causes of evolution by which processes of genetic polymorphisms across species are converted into genetic divergence between species and genetic diversity by investigating the quantity of nucleotides and patterns of nucleotide variation between and within species. Evolutionary mechanisms, such as selection, recombination, mutation, and population structures, are affected by these diversities [23].
An earlier DNA barcoding research of Chenopodium murale produced similar results. According to their findings, the specimen was identified as C. murale using BLAST for both the matK and rbcL genes with 100% sequence matching. Using BOLD, the rbcL gene showed a high degree of similarity to various taxa, including Chenopodium ambrosioides, Chenopodium album, and Chenopodium ficifolium, ranging from 96.3 to 100% [24]. The DNA barcode is clearly distinguish between species, with no overlap between intraspecific and interspecific divergence ideally [25]. Furthermore, the effectiveness of any DNA barcoding approach is determined by the degree of intraspecific and interspecific divergence in a single locus or a set of loci [26]. According to the China Plant BOL Group (2011), the combination rbcL + matK separates 40% of the sampled species in the matched dataset for the Apiaceae family. Furthermore, among all samples, rbcL and matK had 32.1% and 38.6% species identification efficiencies, respectively [27]. According to previous research, both rbcL and matK can be used to identify plants at the species level, because both have a high amplification and sequencing success rate [28]. These two fragments have high identification success rates at the genus and family level [29]. Previous studies reported that genetic distance across accessions could be determined, and that large differences in dissimilarity values could be attributed to genetic differences [30]. For primers matK and rbcL, for instance, grouping based on genetic distance revealed that one group contains different accessions, while the other contains accessions showing wide variations [31]. Plant DNA barcoding is best accomplished by combining coding and noncoding genetic markers. The highly conserved rbcL gene and the more variable matK gene are the most widely explored markers in many researches [32, 33].
In this study, all twelve accessions from different locations of Selinum were examined, and they all belong to the parsley family, Apiaceae. Five-petalled white flower subgenera were easily distinguished based on the anatomical structure of the leaf. Despite the fact that several morpho-anatomical classification methods for the sections and subsections of subgenus Selinum as well as subgenus Angelica have been offered, the links between the subsections and their evolutionary processes are still being contested.
India is a rich source of medicinal plants, and there is no data to identify these plants on molecular basis. Selinum is a medicinal plant with potential therapeutic value. This study has been conducted to identify Selinum using molecular markers because as we go from lower to higher altitude, there are certain changes in morphology and growth within plants which may not be morphologically evident but exist at genetic level. In India, only two species of Selinum, viz S. tenuifolium Wall ex C.B. Clarke and S. vaginatum C. B. Clarke, have been identified on the basis of morphology without any molecular basis ever utilized to authenticate the morphological markers for proper identification of these plants. Various synonyms have been found in Selinum and the ambiguity regarding species with various synonyms. Selinum species are also hard to distinguish morphologically, because certain species have been given different names by different authors instead of a single genuine name. Therefore, DNA barcoding in this plant would be appropriate tool to establish the correct identification of this species or to differentiate species or genus from other members of this family.
The use of morphological features alone is insufficient to adequately define the genus. When investigating interspecific variations, molecular, anatomical, and biochemical markers are more useful than gross morphological ones. A phylogenetic tree can be formed by comparing the same gene sequence across species within a genus, which can support or provide fresh insights into the present taxonomy and rule out the confusion caused by synonymy in Selinum.
Conclusion
The species boundaries of S. tenuifolium and S. vaginatum were assessed using phylogenetic tree using twelve accessions obtained from various places in the Indian Himalayas of Himachal Pradesh and Uttarakhand. The sequencing data collected from the samples used during this study together with other samples retrieved from NCBI were analyzed phylogenetically by using maximum likelihood methods resolved essentially identical topologies. The trees that resulted from this method of analysis provided strong evidence of phylogenetic relationships among the samples investigated in the present study. The samples that were being investigated consistently grouped together forming a monophyletic clade with very strong support, suggesting that the investigated samples and Selinum evolved from a recent common ancestor of Apiaceae family. This finding implies that the genetic divergence in Selinum is phylogenetically informative at the species level. Despite the fact that all of the samples under research were monophyletic, paraphyly was notably noticeable between the samples under investigation and the other Selinum samples obtained from NCBI. In reference to the altitude, the genetic diversity maintained a threshold level of only 3% margins encoded by the comparative analysis of matK and rbcL.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- matK:
-
Maturase K
- rbcL:
-
Ribulose 1,5-bisphosphate carboxylase
- PCR:
-
Polymerase chain reaction
- DNA:
-
Deoxyribonucleic acid
- BLAST:
-
Basic local alignment search tools
- AT:
-
Adenine-thymine
- GC:
-
Guanine-cytosine
- ML:
-
Maximum likelihood
References
Gong X, Sucher NJ (2002) Stroke therapy in traditional Chinese medicine (TCM): prospects for drug discovery and development. Phytomedicine 9(5):478–484. https://doi.org/10.1078/09447110260571760
Marian F, Widmer M, Herren S, Dönges A, Busato A (2006) Physicians’ philosophy of care: a comparison of complementary and conventional medicine. Res Complement Med 13(2):70–77. https://doi.org/10.1159/000090735
Hebert PD, Cywinska A, Ball SL, Dewaard JR (2003) Biological identifications through DNA barcodes. Proc R Soc B: Biol Sci 270(1512):313–321. https://doi.org/10.1098/rspb.2002.2218
Shaw PC, Ngan FN, But PPH, Wang J (1997) Authentication of Chinese medicinal materials by DNA technology. J Food Drug Anal 5:273–284. https://doi.org/10.38212/2224-6614.2926
Tomlinson B, Chan TY, Chan JC, Critchley JA, But PP (2000) Toxicity of complementary therapies: an eastern perspective. J Clin Pharmacol 40:451–456. https://doi.org/10.3349/ymj.2014.55.5.1177
Mitchell A (2008) DNA barcoding demystified. Aust J Entomol 47:169–173. https://doi.org/10.1111/j.1440-6055.2008.00645.x
Moritz C, Cicero C (2004) DNA barcoding: promise and pitfalls. PLoS Biol 2(10):e354. https://doi.org/10.1371/journal.pbio.0020354
Chase MW, Salamin N, Wilkinson M, Dunwell JM, Kesanakurthi RP, Haidar N, Savolainen V (2005) Land plants and DNA barcodes: short-term and long-term goals. Philos Trans R Soc B: Biol Sci 360(1462):1889–1189. https://doi.org/10.1098/rstb.2005.17205
Mallet J, Willmott K (2003) Taxonomy: Renaissance or Tower of Babel. Trends Ecol Evol 18:57–59. https://doi.org/10.1016/S0169-5347(02)00061-7
Schindel DE, Miller SE (2005) DNA barcoding a useful tool for taxonomists. Nature 435:17. https://doi.org/10.1038/435017b
Taberlet P, Coissac E, Pompanon F, Gielly L, Miquel C, Valentini A, Vermat T, Corthier G, Brochmann C, Willerslev E (2007) Power and limitations of the chloroplast trnL (UAA) intron for plant DNA barcoding. Nucleic Acids Res 35(3):e14. https://doi.org/10.1093/nar/gkl938
Hajibabaei M, Dewaard JR, Ivanova NV, Ratnasingham S, Dooh RT, Kirk SL, Mackie PM, Hebert PD (2005) Critical factors for assembling a high volume of DNA barcodes. Philos Trans R Soc B: Biol Sci 360(1462):1959–1967. https://doi.org/10.1098/rstb.2005.1727
Hebert PDN, Gregory TR (2005) The promise of DNA barcoding for taxonomy. Syst Biol 54:852–859. https://doi.org/10.1080/10635150500354886
Pandey MM, Katara A, Pandey G, Rastogi S (2013) Rawat AKS (2013) An important Indian traditional drug of Ayurveda jatamansi and its substitute Bhootkeshi: chemical profiling and antimicrobial activity. Evid Based Complement Alternat Med 2013:142517. https://doi.org/10.1155/2013/142517
Botanical Survey of India (2017) ENVIS centre on floral diversity endemic and threatened taxa. Retrieved March 19, 2018. http://bsienvis.nic.in/Database/E_3942.aspx
Rawat G (2008) Special habitats and threatened plants of India. ENVIS bulletin: wildlife and protected areas. Wildlife Inst India Dehradun 11(1):239 https://www.wii.gov.in/envhome/eindex
The Plant List (2018) Version 1.1. Published on the Internet; Available from: http://www.theplantlist.org/ (Accessed: 07 Jul 2020)
National Center for Complementary and Alternative Medicine (2009) Ayurvedic medicine: an introduction. Retrieved from http://nccam.nih.gov
Hardig TM, Anttila CK, Brunsfeld SJ (2010) A phylogenetic analysis of Salix (Salicaceae) based on matK and ribosomal DNA sequence data. J Bot 197696:12. https://doi.org/10.1155/2010/197696
Chase MW, Cowan RS, Hollingsworth PM, Van Den Berg C, Madriñán S, Petersen G, Seberg O, Jørgsensen T, Cameron KM, Carine M, Pedersen N, Hedderson TAJ, Conrad F, Salazar GA, Richardson JE, Hollingsworth ML, Barraclough TG, Kelly L, Wilkinson M (2007) A proposal for a standardized protocol to barcode all land plants. Taxon 56(2):295–299. https://doi.org/10.1002/tax.562004
Naim DM, Mahboob S (2020) Molecular identification of herbal species belonging to genus Piper within family Piperaceae from northern Peninsular Malaysia. J King Saud Univ Sci 32(2):1417–1426. https://doi.org/10.1016/j.jksus.2019.11.036
Olmstead R, Palmer JD (1994) Chloroplast DNA systematics: a review of methods and data analysis. Am J Bot 81:1205–1224. https://doi.org/10.2307/2445483
Hartl TL, Frost RO (1999) Cognitive-behavioral treatment of compulsive hoarding: a multiple baseline experimental case study. Behav Res Ther 37(5):451–461. https://doi.org/10.1016/s0005-7967(98)00130-2
Ho VT, Tran TKP, Vu TTT et al (2021) Comparison of matK and rbcL DNA barcodes for genetic classification of jewel orchid accessions in Vietnam. J Genet Eng Biotechnol 19:93. https://doi.org/10.1186/s43141-021-00188-1
Chen Y, Zhu X, Loukopoulos P, Weston LA, Albrecht DE, Quinn JC (2021) Genotypic identification of Panicum spp. in New South Wales, Australia using DNA barcoding. Sci Rep 11(1):16055. https://doi.org/10.1038/s41598-021-95610-6
Liu J, Shi L, Han J et al (2014) Identification of species in the angiosperm family Apiaceae using DNA barcodes. Mol Ecol Resour 14(6):1231–1238. https://doi.org/10.1111/1755-0998.12262
Huang X, Ci X, Conran JG, Li J (2015) Application of DNA barcodes in Asian tropical trees - a case study from Xishuangbanna Nature Reserve, southwest China. PLoS One 10:e0129295. https://doi.org/10.1371/journal.pone.0129295
Kang Y, Deng Z, Zang R (2017) DNA barcoding analysis and phylogenetic relationships of tree species in tropical cloud forests. Sci Rep 7:12564. https://doi.org/10.1038/s41598-017-13057-0
Makhadmeh IM, Thabet SG, Ali M (2022) Exploring genetic variation among Jordanian Solanum lycopersicon L. landraces and their performance under salt stress using SSR markers. J Genet Eng Biotechnol 20:45. https://doi.org/10.1186/s43141-022-00327-2
Awad AA (2022) Molecular identification and phylogenetic analysis of Aloe shadensis from Saudi Arabia based on matK, rbcL and ITS DNA barcode sequence. Saudi J Bio Sci 29(2):1125–1133, ISSN 1319-562X. https://doi.org/10.1016/j.sjbs.2021.09.053
Han J, Huang QL, Wu WR, Ma XY, Yang JF, Zhan RT (2016) Molecular identification of six species in Aloe L. based on DNA barcoding. Chin Trad and Herb Drugs 47:1950–1955. https://doi.org/10.7501/j.issn.0253-2670
Yu J, Wu XI, Liu C, Newmaster S, Ragupathy S, Kress WJ (2021) Progress in the use of DNA barcodes in the identification and classification of medicinal plants. Ecotoxicol Environ Saf 208:111691. https://doi.org/10.1016/j.ecoenv.2020.111691
Dong W, Liu J, Yu J, Wang L, Zhou S, Moustafa A (2012) Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS One 7(4):e35071. https://doi.org/10.1371/journal.pone.0035071
Acknowledgements
We are grateful to acknowledge the University Grant Commission, New Delhi (UGC New Delhi, India), for providing financial support to Mr. Ravi Prakash Srivastava (no. 2121430322, ref. no: 21/12/2014). Authors are also thankful to the National Botanical Research Institute (NBRI), Lucknow and Head, Department of Botany, Lucknow University, for providing necessary research facilities.
Funding
This work was supported by the University Grant Commission, New Delhi (UGC New Delhi, India), for providing financial support to Mr. Ravi Prakash Srivastava (grant number 2121430322, ref. no: 21/12/2014).
Author information
Authors and Affiliations
Contributions
RPS designed experiment, collected samples, performed experiments, analyzed data, and wrote the manuscript; LS collected samples, performed experiments, and analyzed data; AS performed experiments; PCV designed experiments and analyzed data; GK performed experiments; and GS designed experiment and analyzed data. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Srivastava, R.P., Saxena, G., Singh, L. et al. Interspecific and intraspecific analysis of Selinum spp. collected from Indian Himalayas using DNA barcoding. J Genet Eng Biotechnol 20, 63 (2022). https://doi.org/10.1186/s43141-022-00345-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43141-022-00345-0