
Abstract
Background
Mastitis is one of the major diseases causing economic loss to the dairy industry by reducing the quantity and quality of milk. Thus, the objective of this scientific study was to find new biomarkers based on genes for the early prediction before its severity.
Methods
In the present study, advanced bioinformatics including hierarchical clustering, enrichment analysis, active site prediction, epigenetic analysis, functional domain identification, and protein docking were used to analyze the important genes that could be utilized as biomarkers and therapeutic targets for mastitis.
Results
Four differentially expressed genes (DEGs) were identified in different regions of the mammary gland (teat cistern, gland cistern, lobuloalveolar, and Furstenberg’s rosette) that resulted in 453, 597, 577, and 636 DEG, respectively. Also, 101 overlapped genes were found by comparing 27 different expressed genes. These genes were associated with eight immune response pathways including NOD-like receptor signaling pathway (IL8, IL18, IL1B, PYDC1) and chemokine signaling pathway (PTK2, IL8, NCF1, CCR1, HCK). Meanwhile, 241 protein-protein interaction networks were developed among overlapped genes. Fifty-seven regulatory events were found between miRNAs, expressed genes, and the transcription factors (TFs) through micro-RNA and transcription factors (miRNA-DEG-TF) regulatory network. The 3D structure docking model of the expressed genes proteins identified their active sites and the binding ligands that could help in choosing the appropriate feed or treatment for affected animals.
Conclusions
The novelty of the distinguished DEG and their pathways in this study is that they can precisely improve the detection biomarkers and treatments techniques of cows’ Escherichia coli mastitis disease due to their high affinity with the target site of the mammary gland before appearing the symptoms.
Graphical abstract

Similar content being viewed by others


Background
Mastitis disease is a parenchymal inflammation of the dairy cattle mammary gland that is one of the most expensive problems in global milk production [1]. In which, mastitis caused significant economic losses for the global dairy industry [2]. The predisposing factors and type of pathogen are important and have a crucial role in the severity and outcome of the complex and multi-etiological nature of the disease, whereas combination of pathogen, environment, host, location, and disease severity makes it more complex and costly [3].
The potential use of bioinformatics analysis for early detection and exploring the pathway’s mechanisms of mastitis infection and its related genes has become one of the hot areas in this field [4]. In which, the advanced bioinformatics models predicted what may happen on the basis of mastitis or even other diseases inflammation. And the most adapted treatment is based on the degree of severity that facilitates early recovery, avoids a significant reduction in reproductive performance, and helps to prevent transmission to other cows. However, little is known about gene articulation in the mammary gland during the infection [5]. Microarray and quantitative real-time PCR techniques have been used to identify genes that become activated in various regions of the mammary gland during the acute period of experimentally induced Escherichia coli mastitis [6, 7]. E. coli infection in mammary tissue shows an increase in TLR2 and TLR4 expression [8]. Mediator and inflammatory cytokine expression are activated by Toll-like receptors (TLRs) which are implicated in cell differentiation, apoptosis, and the immune response. Additionally, the immune response and cytokine’s role in the spread of mastitis has been reported by several literatures [9]. The pro-inflammatory cytokines increase locally and systemically in E. coli-infected glands and mediate the host response via TNF-alpha, IL8, and interleukin 1 beta (IL1B) expression. The transforming growth factor beta induced (TGFBI) and IL-10 cytokines are upregulated during E. coli mastitis, as anti-inflammatory cytokines, and persist for the period of the inflammatory response [10]. A total of 881 differentially expressed genes (DEG) (605 unregulated and 276 downregulated) were distinguished in mammary glands of lactating Chinese Holstein dairy cows compared to dry cows. These genes are responsible for transcription and protein modifications that correlated to mammary gland cell proliferation, cycle, apoptosis, and other immune-related pathways. For instance, RNA-sequencing (RNA-Seq) of the bovine mammary organ at peak lactation of small-tailed sheep (STH) and Gansu Alpine Merino (GAM) provided 74 transcriptome genes profiles that were higher expressed in STH compared with GAM breed. Likewise, 143 genes that were expressed at lower levels in STH compared with GAM were identified. In which, transcriptomic databases, like gene ontogeny (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG), revealed that these DEGs were related to the binding and catalytic activities, hematopoietic cell lineages, oxytocin signaling pathway, and neuroactive ligand-receptor interaction [11].
Furthermore, aggregates of 1103, 1356, and 1397 genes were differentially expressed between Kashmiri and Jersey cattle for multicellular organismal processes and catalytic activities. And these genes were responsible for the majority of the pathways involved in milk production of Jersey dairy cattle, like JAK (Janus tyrosine kinase)-STAT (signal transducer and activator of tranion) (JAK-STAT), p38 MAPK, and the PI3 kinase pathways [11]. A total of 3746 differentially expressed long non-coding RNAs (lncRNAs) (DELs) and 2890 DEGs were distinguished between the dry and lactating mammary glands of Holstein cows. For instance, the functional enrichment analysis on target genes of lncRNAs demonstrated that these genes were involved with lactation-related signaling pathways, including cell cycle, cell adhesion, and phosphatidylinositol-3-kinase (PI3K-Akt) signaling pathways. Furthermore, the interactions among lncRNAs and their potential microRNAs (miRNAs) were anticipated and mostly confirmed [12].
Arora et al. [13] analyzed the milk transcriptome from three lactation phases of Murrah buffalo by RNA sequencing. In that study, a total of 12,833 transcripts were consistent over all the stages and were associated with protein digestion, transport, and immune response. While 271, 205, and 418 were unique to early, mid, and late lactation respectively [13]. As a result, the correlation of the lactation’s various phases may lead to exploring novel genes or transcripts that manage the lactation process. Also, RNA sequencing offers a proficient and far-reaching depiction of the outflow of genes in a given tissue. This process is very important to understand and formulate a therapeutic protocol for bovine mastitis. Therefore, auxiliary research, investigation, and further insights in this area are needed to advance the treatment and disease control in dairy animals. Additionally, various analytical studies have explained the role that E. coli plays in mastitis to elucidate the potential mechanism by which E. coli impairs reproductive performance caused by mastitis in dairy animals. Therefore, specific and sensitive biomarkers for early detection of mastitis to allow early treatment need to be identified and the molecular mechanisms underlying E. coli mastitis need to be better understood.
Vincent et al. [10] mentioned that the local immunization response of dairy cows to E. coli mastitis vaccines has shown limited efficacy because of the modification of the cytokine profile (such as IFN-γ increase and TNF-α reduction). That study results support the idea that the mammary gland cell-mediated immune genes with DEG study links are playing a key role in understanding the E. coli vaccine-induced protection of the mammary gland. Also, in dairy animals infected with coagulated-positive Staphylococci, being in first or second lactation. Several 1700 DEGs were distinguished. The gene ontology functional characterization indicated that the molecular elements of DEGs were overrepresented in the action of cytokines, chemokines, and their receptors. In which, these DEGs are involved in molecular processes, like protein-calcium-lipid binding ability, chemokine activity, and protein homodimerization. Furthermore, gene network analysis has demonstrated that DEGs are related to inflammation, cell relocation, and immune reaction to diseases. Also, they could be responsible for the advancement of cells and tissues, and the cows’ humoral reactions [14].
Thus, this study aimed to find and analyze the insights into the E. coli common pathogenetic factors active in bovine mastitis, and to emphasize the genetic difference among those responses in the mammary gland. Microarray analysis was established to characterize the gene expression patterns and to process their networks that become activated in the bovine mammary regions during the acute phase of experimentally induced intramammary infection with E. coli. The knowledge of the present study could manage the drug discovery related to bovine mastitis through bioinformatics tools assistance for reducing the time and complications involved in the clinical trials related to mastitis disease. In which, the genome-wide analysis of miRNA profile’s relationship with DEG and their target protein active sites provided insight into bovine mastitis and inflammatory diseases in general.
Methods
Data set and description
The profiles of gene expression were downloaded from the Gene expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) with the accession number GSE15441 (platform: GPL3301 USDA Bovine 60mer 344k Array (gene layout) [6]. Three healthy lactating Holstein cows were used to extract four different regions of mammary gland tissues. All the mammary glands were free of any infections. Immediately, following milking in the morning, a 4 ml E. coli suspension (unit of hundred forming colony/ml) was infused into 1 mammary gland of the udder. The contralateral gland was infused with PBS (control gland). After 12 h, the remaining ipsilateral quarter was infused with E. coli. Cows were euthanized for 24 h after the initial infusions and tissues were collected from 4 regions of each mammary gland (Furstenberg’s rosette, cistern, lobuloalveolar, and teat cistern). Twelve samples were also analyzed from the same 4 different regions of mammary glands of 3 Holstein cows from infected and control mammary glands. A total of 24 samples were infused PBS or E. coli after 24 h for the follow-up analysis. Gene expression dataset GSE15020 [15] in gene expression omnibus (GEO), which also contains the mammary gland at 24 h following E. coli infusion in uninfected healthy mammary glands tissue, was used for validation. The data in GSE15020 were produced by the platform of [GPL2112] Affymetrix Bovine Genome Array. A total of 10 samples with two conditions: E. coli treated for 24 h vs. healthy control (five replicates for each condition) were included in this dataset.
Data pre-processing and differentially expressed genes screening
The raw expression data files and the annotation files were downloaded, and the gene expression data of all samples were preprocessed and normalized by using background correction, quantile normalization, and probe summarization using linear models for microarray data (LIMMA) package of Bioconductor (http://www.bioconductor.org/packages/release/bioc/html/limma.html) [16]. The LIMMA package was also used to identify differentially expressed genes (DEGs) between control and infected samples at 24 h post-infection with E. coli from the four different regions under study. The same DEGs screening methods were used in the validation dataset, as well as the cutoff values.
Hierarchical clustering and comparison analysis of selected DEGs in a different region
The expression of the selected DEGs in GSE15441 at 24 h post-infection with E. coli in the four different regions were used to generate hierarchical clustering image by using the P-heatmap package in R tools (RStudio, PBC, Boston, USA) [17]. Then overlapping DEGs (ODEGs) were identified in each group by drawing Venn diagrams using the Venn diagram package in RStudio (https://cran.r-project.org/web/packages/VennDiagram/).
Enrichment analysis for the ODEGs in the four groups
To explore the functions of overlapping differentially expressed genes (ODEGs) and their pathways in E. coli-infected mammary samples, the Database for Annotation, Visualization and Integrated Discovery (DAVID; https://david.ncifcrf.gov/), GO, and KEGG were performed. The P value < 0.05 and gene count ≥ 2 were set as the cut-off criteria. Furthermore, the category of enriched GO, KEGG terms, and the gene number were displayed as a histogram which was constructed by ggplot2 package in R [18].
Protein-protein interaction network analysis for ODEGs
STRING version 10.0 (http://string-db.org/) was used to predict the protein-protein interaction (PPI) relationships of overlapped DEGs (gene pairs with PPI score > 0.4 were used), and Cytoscape 3.2.0 (http://www.cytoscape.org/) were used to construct the PPI network figure.
miRNA-DEGs-TF regulatory network analysis
-
(a)
The miRTarBase database version 2016 (http://mirtarbase.mbc.nctu.edu.tw/) [19] was used to search for miRNAs that target DEGs in the PPI network. The miRNAs obtained from database searching were compared with those reported in the database of cattle candidate genes and genetic markers for milk production and mastitis to find bovine mastitis-related miRNAs.
-
(b)
Searching related transcription factors (TFs) were performed through WebGestalt (http://www.webgestalt.org/option.php) database, which is a functional enrichment analysis tool that can predict the TFs which regulates the ODEGs in PPI network. P value < 0.05 was set as the significance cut-off criteria. Results of (a) and (b) were integrated and miRNA-DEGs-TF regulatory networks were constructed by Cytoscape 3.2.0 (http://www.cytoscape.org/).
Active site prediction of interested DEGs
In biology, the active site is the region of an enzyme where substrate molecules bind and undergo a chemical reaction. Ligands and enzymes are usually bound to the active site of the protein. However, enzyme inhibitors may adhere to the active site and block it. This is very important for the design of enzyme inhibitors. Therefore, we searched active sites of DEGs of interest by using scanprosite (http://prosite.expasy.org/scanprosite/) (Table 1).
Epigenetic analysis, epigenetic modifications, and CpG island
In epigenetic mechanisms, gene nucleotide sequences of cathepsin C (CTSC), IL10, IL8, and IL18 were downloaded from the NCBI database complete with the promoter sequences (1000 bps upstream of the transcription start site), as listed in Table 2. Meth primer 2.0 (http://www.urogene.org/cgi-bin/methprimer/methprimer.cgi) software was used to predict CpG islands within the following parameters: CpG island length > 200 bp, GC content > 50%, observed/expected CpG ratio > 0.6.
Functional domain identification and mutation analysis
Simple Modular Architecture Research Tool (SMART; http://smart.embl-heidelberg.de/) was used for functional domain identification (FDI). Also, the genomic variants database was used to find copy number variants (CNVs) and single-nucleotide polymorphisms (SNPs) of CTSC, IL10, IL8, and IL18 (http://dgv.tcag.ca/dgv/app/about?ref=GRCh37/hg19).
Protein docking exploration of active immune response genes
The 3D structure of 4 selected proteins (CTSC, IL10, IL8, and IL18) that their ligands that play a key role in protein docking were analyzed. Also, protein sequence and structure characterization (α-helix, β-sheet) were performed. In brief, a molecular docking simulation between the key DEGs based on network analysis and the target proteins. Protein Data Bank (PDB, http://www.rcsb.org) was used for downloading the target proteins. The protein structure and active components were modified by AutoDockTools [20, 21]. The processed protein and DEGs were introduced into AutoDock Vina for molecular docking.
Protein sequence-based voluntary
Protein sequence-based voluntary tree to show the homology with other sequences of different species and to find out the similarity of CTSC protein in various species protein sequence alignment was performed. The public database of NCBI (https://www.ncbi.nlm.nih.gov/ncbisearch/) was used to define the protein alignment, the protein sequence of 5 different species under study (Bos taurus, Homo sapiens, Mus musculus, Canis lupus, and Sus scrofa).
Results
Processing of data and screening of differentially expressed genes
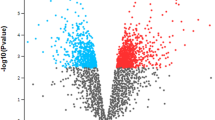
The data were normalized and the detailed normalized expression data can be found in Volcano plots for each region (Fig. 1). In which, only the genes meeting FDR < 0.05 and |log2 fold change (FC)| > 1 were chosen as DEGs. It could be observed that the normalization process controlled the variation between the population’s replicates (Fig S1). Based on the cut-off criteria, the DEGs of 453, 597, 577, and 636 were identified from the teat cistern (Fig. 1a), gland cistern (Fig. 1b), lobuloalveolar (Fig. 1c), and Furstenberg’s rosette (Fig. 1d) regions, respectively.

Volcano plot of DEGs in teat cistern (a), gland cistern (b), lobuloalveolar (c), and Furstenberg’s rosette (d), regions. Red horizontal dot line means FDR = 0.05 cutoff line, two red vertical dot lines mean logFC = 1 and logFC = − 1 cutoff line. Gene dots that were distributed above the FDR = 0.05 red line and out of |logFC| = 1 were the DEGs we identified
Differential Express Genes screening and data processing
The DEG expressions from the four different regions (teat cistern, gland cistern, lobuloalveolar, and Furstenberg’s rosette) were represented by hierarchical clustering heatmaps, as shown in Fig S2. In which, each group was divided into two types (E. coli-infected and control). In the teat cistern, it was observed that E. coli-infected was generally upregulated compared to the control group. However, in Furstenberg’s rosette, the changes in expression pattern between E. coli-infected and control samples were lower. Also, there were 101 overlapped DEGs (ODEG; 100 upregulated and 1 downregulated the gene) (Listed in Table S1). These ODEG were not affected by different tissue regions and were all associated with mastitis (Fig. 2a). Also, the 101 ODEGs had consistent expression patterns in different regions (Fig. 2b).

a Venn diagram of DEGs in the four different regions. b Hierarchical clustering of 101 overlapping differentially expressed genes (ODEGs)
Gene ontology and KEGG pathway analysis for the ODEGs
A total of 27 GO biological processes (BP) for the co-regulated DEGs were generated. In which, 23 of these genes are participating in the immune response, 19 genes with defense response, and 9 genes with inflammatory response. Also, several genes were directly expressed in response to the bacterium (like SLC11A1, IL1B, LBP, IL10) (Table S2). For instance, GO:0006955~immune response gene had the most gene count and FDR with 1.47 × 10−12 (Fig S3). According to KEGG enrichment analysis, ODEGs were significantly enriched in 8 pathways, such as NOD-like receptor signaling pathway (IL8, IL18, IL1B, PYDC1) and the chemokine signaling pathway (PTK2, IL8, NCF1, CCR1, HCK) (Table 1).
Protein-protein interaction network analysis for ODEGs
The STRING database was used to search for PPI relationships among ODEG. In which, 241 PPI pairs were identified with a combined score over than 0.4. The PPI network was constructed, involving 55 upregulated ODEGs (Fig S4). A high significant direct network was found between IL1B and IL8. However, PTK2 was indirectly linked to IL8 through its link with IL1B.
Transcription factor-miRNA-target regulatory network analysis
In total, 57 regulatory relationships between miRNAs, transcription factors (TFs), and ODEGs that had PPI relationships were identified to construct the miRNA-DEG-TF regulatory network. The miRNA-DEG-TF regulatory network contained 21 miRNA-DEG pairs and 36 TF-DEG pairs of s regulatory relationships, including 16 miRNAs obtained from miRTarBase database (Table S3) and 6 significantly enriched TFs obtained from WebGestalt (Table S4). In which, AP1 FDR was calculated 3.59 × 10−5 for the 9 counts of target DEGs: CD79A, FCGR2B, CCR1, MMP3, TREML2, NFATC2, IL10, NCF2, TGM3 (Table S4). Also, two parts of the regulatory relationships were integrated to construct the miRNA-DEG-TF regulatory network. For instance, IL18-DEG was linked with four microRNAs (bta-miR-15b, 16a, 181a, and 155); also, it was integrated with the HNF3-TF (Fig. 3).

miRNA-DEG-TF regulatory network. Red rounded nodes represent DEGs, yellow trigonal nodes represent miRNAs, and blue quadrate nodes represent significantly related TFs; red arrow lines mean TF-DEGs regulatory relationships, green arrow lines mean miRNA-DEGs regulatory relationships
Meanwhile, 5 miRNAs were related to bovine mastitis inflammation from the databases of cattle candidate genes and genetic markers for milk production and mastitis. These miRNAs included bta-miR-146a, bta-miR-21-5p, bta-miR-31, bta-miR-16a, and bta-miR-155. There were 4 DEG that were targeted by the bovine mastitis inflammation-related miRNAs. CTSC was regulated by bta-miR-146a, bta-miR-21-5p, bta-miR-31, as well as was modulated by 3 TF (LEF1, IL10, and IL8). Also, bta-miR-146a and bta-miR-16a, and IL 10 were regulated by 2 transcription factors (LEF1 and AP1). While IL18 was regulated by bta-miR-16a and bta-miR-155, and by HNF3 transcription factor. As a result, the 4 mentioned DEGs that are regulated by bovine mastitis inflammation and that was correlated with miRNAs and TFs were selected for further analysis.
Validation of the expression level of DEGs
In the studied dataset, GSE15441, CTSC, IL10, IL8, and IL18 were upregulated. To validate the expression of the selected 4 DEGs in the GSE15441 dataset, another dataset GSE15020 was used (Fig. 4a). In addition, CTSC, IL10, IL8, and IL18 were all expressed at higher levels (P < 0.05) in E. coli-infected samples than that in healthy samples (Fig. 4b). In which, the expression of IL8 had the highest increase (P < 0.001) compared to IL10. Also, the expression patterns in GSE15020 were the same as GSE15441; therefore, all the 4 selected DEGs (CTSC, IL10, IL8, and IL18) could be validated in GSE15020.

a Heatmap of selected 4 DEGs. b Barplot of selected DEGs. * Refers to 0.01 ≤ p < 0.05, ** refers to 0.005 ≤ p < 0.01, *** refers to p < 0.005. c CpG island in the promoter region of CTST. The blue-filled region is CpG Island. d Presents the active sites in the differentially expressed gene (DEG) of CTSC. e Alignment of CTSC protein among 5 different species. The highlight sequence parts with yellow were consensus sequences of different species
Epigenetic analysis, epigenetic modifications, and CpG island and active cite
Methylation is one of the most important epigenetic modifications. This process includes the addition of methyl groups to the DNA, mostly at CpG sites, to convert cytosine to 5-methylcytosine. As a result, these highly methylated areas tend to be less transcriptionally active. CpG islands were predicted which are the main areas of methylation for the interested genes (gene nucleotide sequences of CTSC, IL10, IL8, and IL18) based on genomic sequences. In this study, CpG islands were found in the promoter region in CTST (from about 500 bp to 900 bp upstream of transcription start site), while no CpG islands were found in IL10, IL8, and IL18 (Fig. 4c). In which, 3 active sites in CTSC were found (Fig. 4d). These 3 active sites were distributed at the 258 aa, 405 aa, and 527 aa positions.
Functional domain identification
We used Simple Modular Architecture Research Tool (SMART, http://smart.embl-heidelberg.de/), which allows the identification and annotation of genetically mobile domains and the analysis of domain architectures. More than 500 domain families found in signaling, extracellular, and chromatin-associated proteins were detected. The results of confidently predicted domains in CTSC, IL10, IL8, and IL18 were listed in Table 3. In which, IL10 domain residues started from 38 to 174; however, CTSC started from 231 to 458. In which, protein sequences alignments from the 5 different species (Bos Taurus, Homo sapiens, Mus musculus, Canis lupus, and Sus scrofa) were compared and highlighted with yellow color to find the consensus sequences of CTSC protein (Fig. 4e). Also, a very intersecting upstream SNP (rs104894210) has been found in CTSC, and two upstream SNPs were in IL8 (rs1000003014, rs1000275946). On the other hand, IL18 had no upstream SNPs (Table S6). In addition, Table S5 listed the CNVs of each gene from the database of Genomic Variants (DGV). In which, novel sequence insertion was in CTSC and two deletion subtypes (nsv1134175, esv2745077) were in IL18.
Protein docking exploration of vibrant immune response genes and the active site prediction of interested DEGs
The 3D structure of the 4 selected proteins (CTSC, IL10, IL8, and IL18) was elucidated and the ligands for docking through protein binding active sites identity are presented in Fig. 5. The active sites of CTSC, IL10, IL8, and IL18 were identified using scanprosite. In which, 3 active sites in CTSC were found by the molecular docking (Fig. 5a, b), but no active sites in IL10, IL8, and IL18 (Fig. 5c, d, e). These 3 active sites were distributed at the 258 aa, 405 aa, and 527 aa positions (Fig. 4d). It has been reported that gene active sites are more easily to be docked by enzymes or ligands, which can regulate the genes’ expression. Therefore, this information is important on target sites for regulatory effects of gene expression based on the controlling of the target genes’ active sites.

3D structure of four selected proteins. Ligand 1 and 2 were ligand molecular which can dock and bind to the active sites of proteins (a, b). Words in the yellow boxes were the information about the active binding sites. In the 3D structures, red ones mean α-helix, the orange ones mean β-fold. a–e Represents CTSC, IL10, IL8, and IL18
Discussion
At present, bovine mastitis is one of the most expensive diseases influencing animal wellbeing. The estimated monetary effect of mastitis in dairy cows on the US economy is 2 billion US dollars of annual lost income [22]. Various microorganisms can cause mastitis, and the sign of the illness can extend from totally subclinical to extreme and hazardous. For instance, Escherichia coli is one of the fundamental pathogens that are responsible for mastitis with its clinical signs in dairy cows. Approximately 5 to 20% of mammary-pathogenic E. coli (MPEC) infections are persistent [22].
Nonetheless, the cure rate of E. coli mastitis is low, and the pathogenesis of E. coli mastitis is not known. Even though the transcriptional reaction of cow mammary gland cells to in vitro infection has been investigated, the interaction and outcomes of these reactions in the in vivo condition of the mammary organ are less clear. Therefore, to generate new procedures and strategies for the rapid diagnosis of E. coli mastitis, an extensive molecular investigation of E. coli mastitis is required. This study coordinated microarray information to distinguish the potential key applicant genes in the dairy cow involved in the pathogenesis of E. coli mastitis [15].
MicroRNAs (miRNAs) assume a pivotal role in controlling inborn and versatile immunity in humans and animals [23]. An aggregate of 1838 miRNAs was recognized, including 580 known-miRNAs (incorporated for the miRbase database) and 1258 anticipated novel miRNAs. The miRNA articulation designs demonstrated that contrasted and control tests, 279 miRNAs and 305 miRNAs were differentially expressed miRNAs (DIE-miRNA) in Staphylococcus aureus and E. coli-infected tissues [24]. In which, Santos et al. [4] concluded that transcriptomics combined with bioinformatics analysis proved the suitability of the discovered drug treatment for recovery from the infectious bovine mastitis by interfering in the mechanisms of action of bovine mammary genes like MTOR and TP53.
In the present study, 4 DEGs in the dataset of GSE15441 were identified from 4 different bovine mammary regions that were infected with E. coli. These DEGs sets shared 101 ODEGs (100 upregulated and 1 downregulated gene), as listed in Table S1. According to the GO BPs and KEGG pathways enrichment analyses, the vast majority of BPs were immune-related, like GO:0006955~immune response, GO:0045087~innate immune response, and GO:0006954~ inflammatory response. Meanwhile, the DEG sets (23 top genes were involved) were significantly (0.05 > P > 0.001) related to the immune response, like IL10, IL8, and IL18. In which, the results confirmed that the immune response was closely related to bovine mastitis. Mandefro et al. [25] mentioned that the use of bovine immunological genes and their polymorphism could be the best disease markers in immunological traits of cattle. In which, a recent study showed that E. coli metabolites are involved in modulating immune functions of its host mammary glands [26]. That mastitis-associated E. coli harbors a higher number of genes for nutrient transport and carbohydrate metabolism through the TCA cycle [27]. The methyl-accepting chemotaxis proteins (MCP) or genes (tar, tsr, and tap) act through transmembrane receptors that mediate chemotactic response and cell-to-cell communication of E. coli [27]. The activation of the tumor repressor protein, p53, appears to play a central role in modulating the expression of various mammary epithelial cells involution responsive genes [28]. Thus, at the molecular level of the mammary gland immune response during infection by pathogenic bacteria, it is fundamental to design an effective therapy to control and eradicate bovine mastitis [29]. Therefore, the uniqueness of DEGs is that they are participating in the immune response-related BPs to genetically control the bovine mastitis problem. Among the 23 genes which participated in the immune response, IL8, IL18, IL1B, and PYDC1 were identified as being significant in the NOD-like receptor signaling pathway. These NOD-like receptors play an important role in host defense against the infection with E. coli mastitis [30]. Impairment of the NOD1/NF-κB pathway tends to downregulate mRNA levels of chemokine IL-8 [31]. As we further narrowed the scope of the important DEGs by finding the most important genes (CTSC, IL10, IL8, and IL18) (Fig. 3) [32]. In which, the integration between these DEGs and the reported bovine mastitis related miRNAs bta-miR-146a [33], bta-miR-21-5p, bta-miR-31 [5], bta-miR-16a, and bta-miR-155 [14] were done (Fig. 3). Furthermore, CTSC, IL10, IL8, and IL18 were verified to have the same expression pattern in GSE15441 and in GSE15202 [34]. In which, it suggests that their over-expression patterns in mammary tissue were not accidental but from the tendency of the representative biological targets.
Conclusions
In this study, 2263 DEGs aggregates were identified in the different regions of the mammary gland (teat cistern, gland cistern, lobuloalveolar, and Furstenberg’s rosette). That resulted in 453, 597, 577, and 636 DEGs, respectively. Through bioinformatics investigation, IL8, IL10, IL18, and CTSC were recognized as the essential biomarkers for E. coli mastitis and were related to the immune response of bovine mastitis. This study provides potential key information for using the target genes as biomarkers of early cattle mastitis diagnosis. Also, further integrating of the DEGs and the TF that related to the immune response of the infecting pathogens was established to facilitate the development of the target drugs for controlling the transcription pathways of the infected animals against E. coli mastitis.
Availability of data and materials
All the data and their availability are included in the manuscript text, figures, tables, and supplementary materials. Also, all relevant raw data will be freely available to any scientist wishing to use them for non-commercial purposes, without breaching.
Abbreviations
- DEG:
-
Differentially expressed genes
- ODEG:
-
Overlapping differentially expressed genes
- IL:
-
Interleukin
- IL1B:
-
Interleukin 1 beta
- CTSC:
-
Cathepsin C
- PYDC1:
-
Pyrin domain-containing protein 1
- PTK:
-
Protein tyrosine kinase
- NCF:
-
Neutrophil cytosolic factor
- CCR:
-
C-C motif chemokine receptor
- HCK:
-
Hemopoetic cell kinase
- miRNAs:
-
Micro ribonucleic acid
- TFs:
-
Transcription factors
- 3D:
-
Three dimension
- E. coli :
-
Escherichia coli
- TLR:
-
Toll-like receptor
- TGFBI:
-
Transforming growth factor beta induced
- RT-PCR:
-
Reverse transcriptase polymerase chain reaction
- STH:
-
Small-tailed sheep
- GAM:
-
Gansu Alpine Merino
- GO:
-
Gene ontogeny
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- JAK-STAT:
-
JAK (Janus tyrosine kinase)-STAT (signal transducer and activator of tranion)
- MAPK:
-
Mitogen-activated protein kinase
- PI3K-Akt:
-
Phosphatidylinositol-3-kinase
- GEO:
-
Gene expression omnibus
- GSE:
-
Genetics selection evolution
- LIMMA:
-
Linear models for microarray data
- DAVID:
-
Database for Annotation Visualization and Integrated Discovery
- PPI:
-
Protein-protein interaction
- SMART:
-
Simple Modular Architecture Research Tool
- SNP:
-
Single-nucleotide polymorphism
- CNV:
-
Copy number variants
References
Adriaens I, Van Den Brulle I, Geerinckx K et al (2021) Milk losses linked to mastitis treatments at dairy farms with automatic milking systems. Prev Vet Med 194:105420. https://doi.org/10.1016/J.PREVETMED.2021.105420
Barkema HW, von Keyserlingk MAG, Kastelic JP, Lam TJGM, Luby C, Roy JP, LeBlanc SJ, Keefe GP, Kelton DF (2015) Invited review: changes in the dairy industry affecting dairy cattle health and welfare. J Dairy Sci 98(11):7426–7445. https://doi.org/10.3168/JDS.2015-9377
De Vliegher S, Fox LK, Piepers S et al (2012) Invited review: mastitis in dairy heifers: nature of the disease, potential impact, prevention, and control. J Dairy Sci 95(3):1025–1040. https://doi.org/10.3168/jds.2010-4074
Sobrinho Santos EM, Almeida AC, Santos HO, Cangussu AR, Costa KS, Alves JN, Bertucci Barbosa LC, Souza Aguiar RW (2019) Mechanism of Brassica oleracea performance in bovine infectious mastitis by bioinformatic analysis. Microb Pathog 129:19–29. https://doi.org/10.1016/J.MICPATH.2019.01.029
Pathak S, Grillo AR, Scarpa M, Brun P, D’Incà R, Nai L, Banerjee A, Cavallo D, Barzon L, Palù G, Sturniolo GC, Buda A, Castagliuolo I (2015) MiR-155 modulates the inflammatory phenotype of intestinal myofibroblasts by targeting SOCS1 in ulcerative colitis. Exp Mol Med 47(5):e164. https://doi.org/10.1038/emm.2015.21
Rinaldi M, Li RW, Bannerman DD, Daniels KM, Evock-Clover C, Silva MVB, Paape MJ, van Ryssen B, Burvenich C, Capuco AV (2010) A sentinel function for teat tissues in dairy cows: dominant innate immune response elements define early response to E. coli mastitis. Funct Integr Genomics 10(1):21–38. https://doi.org/10.1007/s10142-009-0133-z
Dai W-T, Zou Y-X, White RR et al (2018) Transcriptomic profiles of the bovine mammary gland during lactation and the dry period. Funct Integr Genomics 18(2):125–140. https://doi.org/10.1007/S10142-017-0580-X/TABLES/1
Mitterhuemer S, Petzl W, Krebs S et al (2010) Escherichia coli infection induces distinct local and systemic transcriptome responses in the mammary gland. BMC Genomics 11:1–16
Miles AM, Huson HJ (2021) Graduate student literature review: understanding the genetic mechanisms underlying mastitis. J Dairy Sci 104(1):1183–1191. https://doi.org/10.3168/jds.2020-18297
Herry V, Gitton C, Tabouret G et al (2017) Local immunization impacts the response of dairy cows to Escherichia coli mastitis. Sci Rep 7(1):3441. https://doi.org/10.1038/S41598-017-03724-7
Hao Z, Zhou H, Hickford JGH, Gong H, Wang J, Hu J, Liu X, Li S, Zhao M, Luo Y (2019) Transcriptome profile analysis of mammary gland tissue from two breeds of lactating sheep. Genes (Basel) 10(10):781. https://doi.org/10.3390/genes10100781
Yang B, Jiao B, Ge W et al (2018) Transcriptome sequencing to detect the potential role of long non-coding RNAs in bovine mammary gland during the dry and lactation period. BMC Genomics 19:1–14
Arora R, Sharma A, Sharma U et al (2019) Buffalo milk transcriptome: a comparative analysis of early, mid and late lactation. Sci Rep 9:1–9
Kosciuczuk EM, Lisowski P, Jarczak J et al (2017) Transcriptome profiling of Staphylococci-infected cow mammary gland parenchyma. BMC Vet Res 13:1–12
Li L, Chen X, Chen Z (2019) Identification of key candidate genes in dairy cow in response to escherichia coli mastitis by bioinformatical analysis. Front Genet 10:1251. https://doi.org/10.3389/fgene.2019.01251
Wang L, Cao C, Ma Q et al (2014) RNA-seq analyses of multiple meristems of soybean: novel and alternative transcripts, evolutionary and functional implications. BMC Plant Biol 14:1–19
Fox J, Weisberg S (2011) An R companion to applied regression. Sage, Thousand Oaks
Wickham H (2011) ggplot2. Wiley Interdiscip Rev Comput Stat 3(2):180–185. https://doi.org/10.1002/wics.147
Chou C-H, Chang N-W, Shrestha S, Hsu SD, Lin YL, Lee WH, Yang CD, Hong HC, Wei TY, Tu SJ, Tsai TR, Ho SY, Jian TY, Wu HY, Chen PR, Lin NC, Huang HT, Yang TL, Pai CY, Tai CS, Chen WL, Huang CY, Liu CC, Weng SL, Liao KW, Hsu WL, Huang HD (2016) miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res 44(D1):D239–D247. https://doi.org/10.1093/nar/gkv1258
Gao K, Song Y-P, Song A (2020) Exploring active ingredients and function mechanisms of Ephedra-bitter almond for prevention and treatment of corona virus disease 2019 (COVID-19) based on network pharmacology. BioData Min 13(1):1–20. https://doi.org/10.1186/S13040-020-00229-4
Wang L, Wang Z, Yang Z, Yang K, Yang H (2021) Study of the active components and molecular mechanism of Tripterygium wilfordii in the treatment of diabetic nephropathy. Front Mol Biosci 8:1. https://doi.org/10.3389/FMOLB.2021.664416
Lippolis JD, Holman DB, Brunelle BW, Thacker TC, Bearson BL, Reinhardt TA, Sacco RE, Casey TA (2018) Genomic and transcriptomic analysis of Escherichia coli strains associated with persistent and transient bovine mastitis and the role of colanic acid. Infect Immun 86(1):1–12. https://doi.org/10.1128/iai.00566-17
Farid EA, Gouda MM, Hussein LA, Ahmed N et al (2017) Role of melt curve analysis in interpretation of nutrigenomics’ microRNA expression data. Cancer Genomics Proteomics 14(6):469–481. https://doi.org/10.21873/cgp.20057
Luoreng Z-M, Wang X-P, Mei C-G, Zan L-S (2018) Comparison of microRNA profiles between bovine mammary glands infected with Staphylococcus aureus and Escherichia coli. Int J Biol Sci 14(1):87–99. https://doi.org/10.7150/ijbs.22498
Mandefro A, Sisay T, Kim K-S, Edea Z, Konwarh R, Dadi H (2021) Single nucleotide polymorphisms of leptin gene in five Ethiopian indigenous cattle breeds and the Korean Hanwoo breed. Trop Anim Heal Prod 53(2):1–11. https://doi.org/10.1007/S11250-021-02642-1
Hoque MN, Istiaq A, Rahman MS, Islam MR, Anwar A, Siddiki AMAMZ, Sultana M, Crandall KA, Hossain MA (2020) Microbiome dynamics and genomic determinants of bovine mastitis. Genomics 112(6):5188–5203. https://doi.org/10.1016/J.YGENO.2020.09.039
Li N, Wang Y, You C, Ren J, Chen W, Zheng H, Liu Z (2018) Variation in raw milk microbiota throughout 12 months and the impact of weather conditions. Sci Rep 8(1):1–10. https://doi.org/10.1038/s41598-018-20862-8
Nobrega DB, Naushad S, Naqvi SA et al (2018) Prevalence and genetic basis of antimicrobial resistance in non-aureus Staphylococci isolated from Canadian dairy herds. Front Microbiol 9. https://doi.org/10.3389/FMICB.2018.00256
Oviedo-Boyso J, Valdez-Alarcón JJ, Cajero-Juárez M, Ochoa-Zarzosa A, López-Meza JE, Bravo-Patiño A, Baizabal-Aguirre VM (2007) Innate immune response of bovine mammary gland to pathogenic bacteria responsible for mastitis. J Infect 54(4):399–409. https://doi.org/10.1016/j.jinf.2006.06.010
Sharun K, Dhama K, Tiwari R et al (2021) Advances in therapeutic and managemental approaches of bovine mastitis: a comprehensive review. Vet Q 41(1):107–136. https://doi.org/10.3168/jds.2020-18297
Wei L-J, Tan X, Fan G-J, Jiang YN, Shah QA (2016) Role of the NOD1/NF-κB pathway on bovine neutrophil responses to crude lipopolysaccharide. Vet J 214:24–31. https://doi.org/10.1016/j.tvjl.2016.02.006
Lai Y-C, Fujikawa T, Maemura T, Ando T, Kitahara G, Endo Y, Yamato O, Koiwa M, Kubota C, Miura N (2017) Inflammation-related microRNA expression level in the bovine milk is affected by mastitis. PLoS One 12(5):e0177182. https://doi.org/10.1371/journal.pone.0177182
Jin W, Ibeagha-Awemu EM, Liang G et al (2014) Transcriptome microRNA profiling of bovine mammary epithelial cells challenged with Escherichia coli or Staphylococcus aureus bacteria reveals pathogen directed microRNA expression profiles. BMC Genomics 15:1–16
Jensen K, Günther J, Talbot R et al (2013) Escherichia coli-and Staphylococcus aureus-induced mastitis differentially modulate transcriptional responses in neighbouring uninfected bovine mammary gland quarters. BMC Genomics 14:1–19
Acknowledgments
The authors are grateful for the financial assistance by the project Research Platform Construction of the Ministry of Agriculture International Exchange and Cooperation in Science and Technology and the Opening Project of Guangxi Key Laboratory of Buffalo Genetics, Reproduction and Breeding (SNKF-2016-01).
Funding
This study was funded by the Project Research Platform Construction of the Ministry of Agriculture International Exchange and Cooperation in Science and Technology and the Opening Project of Guangxi Key Laboratory of Buffalo Genetics, Reproduction and Breeding (SNKF-2016-01). The funder supported the experiment data analyses and software fees.
Author information
Authors and Affiliations
Contributions
FU, XL, and SZ conceived and designed the experiments. FU, MG, NMS, MS, and MK wrote the original draft of the manuscript. ZS and XL were the major supervisors. FAK, ZUR, and HST helped in data analysis. MG and NMS helped in manuscript reviewing and editing, and overall critical review of the manuscript. Also, all authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
The authors declare that the work described was an original manuscript that followed all the ethical regulations and that has not been published previously. Also, no animal or human trails were constructed and no conflict of interest exists in the submission of this manuscript, and the manuscript is approved by all authors to be published.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig S1.
(a) Boxplot of GSE15441 data preprocessing before and after normalization. (b) Boxplot of GSE15020 data preprocessing before and after normalization. Blue and grey boxes refer to E. coli infected and healthy mammary tissue samples. Fig S2. Hierarchical clustering of DEGs in (a) teat cistern, (b) gland cistern, (c) lobuloalveolar, and (d) Furstenberg’s rosette. Fig S3. (a) The histogram of the category of enriched GO BP terms for the 101 ODEGs. (b) The histogram of the category of enriched KEGG pathways for the 101 Overlapping Differentially Express Genes (ODEGs). The horizontal axis represents the number of genes, and the vertical axis represents terms. The color bar means changes of significance. Fig S4. PPI network of Overlapping Differentially Express Genes (ODEGs). Table S1. Presents the 101 overlapped DEGs (ODEG; 100 up-regulated and 1 down-regulated the gene). Table S2. Enriched GO BPs for 101 ODEGs. Table S3. List of searched miRNAs from the miRTarBase database. Table S4. List of searched TFs from WebGestalt. Table S5. Copy Number Variants (CNVs) of CTSC, IL10, IL8, and IL18. Table S6. Single nucleotide polymorphisms (SNPs) of CTSC, IL10, IL8, and IL18.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Farmanullah, F., Liang, X., Khan, F.A. et al. Transcriptomic in silico analysis of bovine Escherichia coli mastitis highlights its immune-related expressed genes as an effective biomarker. J Genet Eng Biotechnol 19, 153 (2021). https://doi.org/10.1186/s43141-021-00235-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43141-021-00235-x