
Abstract
Background
Targeting viral cell entry proteins is an emerging therapeutic strategy for inhibiting the first stage of SARS-CoV-2 infection. In this study, 106 bioactive terpenoids from African medicinal plants were screened through molecular docking analysis against human angiotensin-converting enzyme 2 (hACE2), human transmembrane protease serine 2 (TMPRSS2), and the spike (S) proteins of SARS-CoV-2, SARS-CoV, and MERS-CoV. In silico absorption-distribution-metabolism-excretion-toxicity (ADMET) and drug-likeness prediction, molecular dynamics (MD) simulation, binding free energy calculations, and clustering analysis of MD simulation trajectories were performed on the top docked terpenoids to respective protein targets.
Results
The results revealed eight terpenoids with high binding tendencies to the catalytic residues of different targets. Two pentacyclic terpenoids (24-methylene cycloartenol and isoiguesteri) interacted with the hACE2 binding hotspots for the SARS-CoV-2 spike protein, while the abietane diterpenes were found accommodated within the S1-specificity pocket, interacting strongly with the active site residues TMPRSS2. 3-benzoylhosloppone and cucurbitacin interacted with the RBD and S2 subunit of SARS-CoV-2 spike protein respectively. These interactions were preserved in a simulated dynamic environment, thereby, demonstrating high structural stability. The MM-GBSA binding free energy calculations corroborated the docking interactions. The top docked terpenoids showed favorable drug-likeness and ADMET properties over a wide range of molecular descriptors.
Conclusion
The identified terpenoids from this study provides core structure that can be exploited for further lead optimization to design drugs against SARS-CoV-2 cell-mediated entry proteins. They are therefore recommended for further in vitro and in vivo studies towards developing entry inhibitors against the ongoing COVID-19 pandemic.
Similar content being viewed by others


Background
The coronavirus disease-19 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was declared a public health emergency by the World Health Organization (WHO) [8, 55, 66, 67]. The death toll from this virus has by far surpassed that of 2003 severe acute respiratory syndrome-coronavirus (SARS-CoV) and the 2012 Middle East respiratory syndrome coronavirus (MERS-CoV) outbreaks combined [21, 53]. The SARS-CoV-2 earlier known as 2019 novel coronavirus (2019-nCoV) is evolutionarily related (80% identity) to SARS-CoV [9]. It causes multiple organ failures, which may present as fever, cough, shortness of breath, dyspnea, pneumonia, severe acute respiratory syndrome, kidney failure, and even death [31, 68]. Bioinformatics has proven a notable tool in understanding the virulence and interaction of the SARS-CoV-2 to different receptors [32, 56, 57]. Cell entry of coronaviruses depends on a fine interplay between the viral membrane spike (S) proteins and the host cell membrane proteins more importantly are the angiotensin-converting enzyme 2 (ACE2) and serine protease transmembrane protease serine 2 (TMPRSS2) [7]. The S-protein comprises two subunits; S1 as the receptor-binding domain (RBD) while S2 subunit is for the fusion of viral membrane and host cellular membrane. The SARS-CoV-2 relies on the host ACE2 for entry and the TMPRSS2 for S-protein priming. Upon binding of the S-protein to host receptor through the receptor-binding domain (RBD) in the S1 subunit, the S2 subunit mediates fusion of the viral envelope with the host membranes [12]. Although the overall sequence similarity between S-protein of SARS-COV-2 and SARS-CoV is approximately ~ 76%, affinity between S-RBD of SARS-COV-2 and ACE2 is found to be approximately four fold higher when compared with SARS-CoV RBD [12, 64]. This molecular interaction is responsible for regulating both the cross-species and higher human-to-human transmissions of SARS-CoV-2 [63, 74]. Therefore, these protein effectors of viral attachment, membrane fusion, and cell entry are known as emerging targets for development of entry inhibitors, antibodies, and vaccines [74].
The use of phytomedicines as alternatives to combat viral diseases and other infections forms an integral component of African cultural practices, and hence a prominent feature in Africa [3, 5, 18, 37, 41, 61]. Terpenoids are a well-known class of phytochemicals of tremendous pharmaceutical value over time because of their relevant broad-spectrum utility in medicine [17, 23, 40]. Screening a database of phytochemicals from indigenous African medicinal plants may help identify terpenoids with therapeutic potentials against the COVID-19 pandemic. Therefore, this study explores computational screening of terpenoids from indigenous African medicinal plants as potential inhibitors of the emerging proteins responsible for coronavirus cell entry and subsequent infection.
Methods
Protein preparation
The crystal structures of proteins for the docking studies were retrieved from the Protein Databank (http://www.rcsb.org) with their various PDB identification codes [1R42: angiotensin-converting enzyme 2 (ACE2) [58]; 2OQ5: type II transmembrane serine proteinases (TMPRSS2) [26]; 6vw1: 2019-nCoV chimeric receptor-binding domain complexed with its receptor human ACE2 (ACE2-RBD) [49] and coronaviruses spike protein (6VSB: SARS-CoV-2) [67]; (5X5B: SARS-CoV) [72] and (5x5c: MERS-CoV) [72]. All the crystal structures were prepared by removing existing ligands and water molecules, missing hydrogen atoms were added while the Kollamn charge were added as the partial atomic charge using MGL-AutoDockTools (ADT, v1.5.6) [36]. The well-ordered scheme was repeated for each protein and thereafter saved into dockable pdbqt format for molecular docking.
Ligand preparation
One hundred and six bioactive terpenoids from African medicinal plants were collected based on literature search. Structure Data Format (SDF) of the reference inhibitors (S1: MLN-4760; S2: camostat and S3: nelfinavir mesylates) and 106 bioactive terpenoids derived from African plants were retrieved from the PubChem database (www.pubchem.ncbi.nlm.nih.gov) and converted to mol2 chemical format using Open babel [39]. Other compounds that were not available on the database were drawn with Chemdraw version 19 and converted to mol2 chemical format. Polar hydrogen charges of the gasteiger-type were assigned and the nonpolar hydrogen molecules were merged. The ligand molecules were further converted to the dockable pdbqt format using MGL-AutoDockTools (ADT, v1.5.6) [36].
Molecular docking
Molecular docking was performed to evaluate the binding affinity and to provide initial coordinates and topology parameters for the MD simulations. The screening of human enzymes and active regions of the coronaviruses spike protein and determination of binding affinities were carried out using AutoDock Vina [59]. The binding scores from vina analysis were further validated by BINDSURF [48]. Docking of bioactive terpenoids and reference compounds against human ACE2, human TMPRSS2, and SARS-CoV-2 spike protein was performed by AutoDock Vina to locate alternate binding sites enclosing the whole macromolecules. Default settings of Vina wase used, as the scoring matrix in this program is stochastic, and each run uses a random seed position except for the grid box which was adjusted with extended grid size (60 Å × 60 Å × 60 Å) to reveal all the possible interaction sites. The molecular docking was executed using a flexible docking protocol; all bonds contained in ligand were allowed to rotate freely, making the receptor rigid. Once the molecular docking experiments were completed and 10 configurations for each protein-ligand complex were generated for all the terpenoids, text files of scoring results were also produced for the purpose of manual comparative analysis. The top docked terpenoids were uploaded into the respective columns of BINDSURF webserver to validate and calculate the energetic interactions. The molecular interactions between proteins and selected compounds with higher binding affinity to the proteins were viewed with Discovery Studio Visualizer version 16.
Molecular dynamics simulation
Molecular dynamics simulations were carried out on the top ranked terpenoid to respective protein targets (SARS-CoV-2 spike (S) protein, human angiotensin-converting enzyme 2 (ACE2), and transmembrane protease serine 2 (TMPRSS2)). The complexes were prepared and solvated, in TIP3P water model and neutralized by adding NaCl ions and its concentration was set to 0.154 M using CHARMM-GUI webserver prior to running MD simulation using Nanoscale Molecular Dynamics (NAMD V 2.13) [6, 27, 44]. The ligands (terpenoids) were parameterization on the SwissParam webserver. The TIP3P water model was used to resemble the added water box, with 10 Å padding, for the periodic boundary condition to be applied [34]. Nose-Hoover Langevin piston was used to control the pressure at 1.01325 bar. In contrast, Langevin dynamics controlled the system’s temperature at the physiological value (310 K, 7.0, and 0.154 M NaCl, respectively). The time step was set at its default two fs with SHAKE approximation. Visualizing molecular dynamics (VMD 1.9.3) software was used to prepare the input files and analyze the output trajectories [22]. Minimization step for the complexes was initiated for 10,000 steps using a conjugate gradient algorithm in constant number of atoms, constant volume, and constant temperature ensemble (NVT) using CHARMM 36 force field. Afterwards, equilibration of each system for one nanosecond was started in constant number of atoms, constant pressure, and constant temperature ensemble (NPT). Finally, a production run for 100 ns for each system was initialized in NVT ensemble. Periodic Boundary Conditions (PBC) was applied to the simulation. Trajectories were extracted each 0.1 ns and time step was set to 2 femto second. The analysis of the dynamics was performed by utilizing VMD scripts to calculate root mean square deviation (RMSD), root mean square fluctuation (RMSF), surface accessible surface area (SASA), radius of gyration (RoG), and hydrogen bonds (H-bonds) [22]. All the analyses were performed after removing the PBC using the pbctools package in VMD using this command pbc unwrap-sel “selection” where selection is replaced by the appropriate name.
Clustering of molecular dynamic trajectory
Afterwards, TTClust V 4.9.0 [60] was used to cluster the whole trajectory (1000 frame) using the elbow method to calculate the optimum number of clusters. For each representative frame produced, Protein Ligand Interaction Profiler (PLIP) [47] was used to know the types and number of interactions between the protein and the ligand.
MM/GBSA calculation and MM/GBSA free energy decomposition analysis
To calculate the binding free energies of the top docked terpenoids to each of the protein target, molecular mechanics–generalized born surface area (MM-GBSA) was calculated using the version implemented in AmberTools 20 for all frames in the trajectory [35, 54]. Saltcon variable was set to 0.154 M and igb, which determines the generalized born method to use, was set to the default value of five. After the decomposition process, the energy contribution could be distributed to each residue of receptor and the binding interaction of each ligand-residue pair consists of three energy terms: van der Waals contribution (ΔEvdw), electrostatic contribution (ΔEele), and the desolvation term (ΔGdesolvation) which included the polar (ΔGGB), the non-polar (ΔGSA), and total free energy (∆Gtotal) term. Fifty frames separated by equal intervals of 20 frames were used to generate the binding free energies and were also used for the free energy decomposition analysis.
Drug-likeness and ADMET studies
The top terpenoids that demonstrated highest binding affinity for ACE2, TMPRSS2, and active regions of SARS-CoV-2 spike protein were subjected to several drug-likeness predictive descriptors which orally bio-active drug should comply [30, 38]. The predicted absorption, distribution, metabolism, excretion, and toxicity (ADMET) studies were analyzed using the ADMET webserver [10]. The SDF file and SMILES of the compounds were downloaded from PubChem database to calculate ADMET properties using default parameters.
Results
Molecular docking
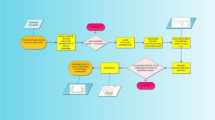
Figure 1 provides a flow chart showing the stepwise screening of African derived terpenoids for potential inhibitors of SARS-CoV-2 cell entry proteins.

Flow chart showing the stepwise screening of African derived terpenoids for potential inhibitors of membrane-mediated SARS-CoV-2 cell entry
The result from the docking analysis of the reference inhibitors and bioactive terpenoids with the human ACE2, TMPRSS2, and SARS-CoV-2 spike protein is shown in Table S1 (supplementary material). The top 20 terpenoids with the highest binding affinity for the ACE2 were further analyzed for binding interactions with SARS-CoV-2 chimeric receptor-binding domain complexed with its human receptor ACE2 (ACE2-RBD) and the S protein of SARS-CoV and MERS-CoV (Table S3, supplementary material) (Fig. 2).

Chemical structure of terpenoid with remarkable binding energy to human ACE2, TMPRSS2, and SARS-coronaviruses S protein (T1) 24-methylene cycloartenol; (T2) Isoiguesterin; (T3) 11-hydroxy-2-(3,4-dihydroxybenzoyloxy)abieta-5,7,9(11),13-tetraene-12-one; (T4) 11-Hydroxy-2-(4-hydroxybenzoyloxy)-abieta-5,7,9(11),13-tetraene-12-one; (T5) 3-benzoylhosloppone; (T6) Cucurbitacin B; (T7) 7-deacetoxy-7-oxogedunin and (T8) 3-Friedelanone
The docking analysis revealed that the reference inhibitor (MLN-4760) to the human ACE2 protein had binding energy of − 7.7 Kcal/mol, respectively, while camostat an inhibitor of TMPRSS2 had a binding energy of − 7.6 Kcal/mol as represented in Fig. 3. It was further observed that the topmost docked terpenoids to the ACE2 had higher binding affinity for the S protein of SARS-CoV and MERS-CoV than SARS-CoV-2. More than 10 terpenoids had higher binding affinity than the 3 inhibitors used in this study (Table S1: supplementary material). The top 20 docked compounds to SARS-CoV-2 S-proteins had higher binding affinity than nelfinavir mesylates (Table S3: Supplementary material).

AutoDock binding energies (Kcal/mol) of reference inhibitors and top bioactive terpenoids with human angiotensin-converting enzyme 2 (ACE2), transmembrane protease serine 2 (TMPRSS2), ACE2-SARS-CoV-2 spike receptor binding domain complex (ACE2-RBD), and (*S P) spike protein of coronaviruses. S1 MLN-4760. S2 Camostat. S3 Nelfinavir mesylates. T1 24-methylene cycloartenol. T2 Isoiguesterin. T3 11-hydroxy-2-(3,4-dihydroxybenzoyloxy)abieta-5,7,9(11),13-tetraene-12-one. T4 11-hydroxy-2-(4-hydroxybenzoyloxy)-abieta-5,7,9(11),13-tetraene-12-one. T5 3-benzoylhosloppone. T6 Cucurbitacin B. T7 7-deacetoxy-7-oxogedunin. T8 3-Friedelanone
From the binding scores generated by the interacting terpenoids with the ACE2 and TMPRSS2 proteins, the top 2 docked terpenoids with the highest binding affinity are 24-methylene cycloarteno and isoiguesterin with corresponding binding energy of − 9.7, and − 9.5 Kcal/mol, respectively. The best two docked terpenoids to SARS-CoV-2 S protein are 3-benzoylhosloppone and cucurbitacin with binding energies of − 9.4 and − 9.3 Kcal/mol respectively. 3-benzoylhosloppone had the highest binding affinity for SARS-CoV-2 S protein and the second top binding affinity to MERS-CoV S protein (Fig. 3).
Interaction of selected terpenoids with amino acids of target proteins
The amino acid interactions of the human target proteins (ACE2 and TMPRSS2) with reference inhibitors and plant derived terpenoids that demonstrated the highest binding tendencies are represented in Table 1. In the same way, the amino acid residues of the coronaviruses S protein that interacted with reference inhibitors and terpenoids with the highest binding affinity are shown in Table 2. The interacting residues of human ACE2 and TMPRSS2 with respective ligand groups were majorly through hydrophobic interactions and H-bond. Few H-bonding below 3.40 Å were observed with coronaviruses S protein (Table 1 and Fig. 3). The binding of MLN-4760 to ACE2 showed that it was docked into the N terminus and zinc-containing subdomain I of ACE2 (Fig. 4a). MLN-4760 exhibited several types of hydrophobic interactions (Pi-Sigma, Pi-Pi T-Shaped, Pi-Alkyl, and Alkyl) with TYR510 PHE504 MET360, LYS363, and CYS344, a salt and attractive charges to ARG514, ARG518, and ARG278 and hydrogen bond to TYR515, THR371, PRO346, and ARG273 (Fig. 4a). 24-methylene cycloartenol the best docked terpenoid was docked into the C terminus-containing subdomain II of ACE2 but interacted with different residue as with the case of N-acetyl-D-glucosamine (Fig. 4b). 24-methylene cycloartenol interacted via H-bond to TRP163, SER170, and TYR497. A Pi-Alkyl interaction was also observed with TYR613, PRO492, and VAL491. Isoiguesterin interacted via H-bond to ASP350, TYR385, and ASN394. A Pi-Alkyl and Alkyl interactions was observed with the ALA99, PHE40, PHE390, LEU73, and TRP69 residues respectively in a similar binding pattern with MLN-4760 (Fig. 4c). Camostat was docked into the S1-specificity pocket of TMPRSS2 (Fig. 5a). It interacted via conventional H-bond to five amino residues (ARG41, SER195, TRP215, ALA190, and ASP189) and via carbon hydrogen bond to GLN192 of TMPRSS2. The conventional H-bond was formed in the direction of the guanidine group in this order: first ester bond, second ester bond, while the last three residues interacted with amidino nitrogen of guanidine group, respectively. The phenyl ring was responsible for the carbon-hydrogen bond with GLN192 (Fig. 5a). T3 and T4 were docked into S1-specificity pocket of TMPRSS2 in a similar binding pattern as in the case of camostat (Fig. 5b, c). The only difference observed between the binding pattern of T3 and T4 was an additional H-bond between T3 with ARG41 (Fig. 5b). Nelfinavir mesylates an inhibitor of SARS-CoV and MERS-CoV S protein interacted in its best docked conformation to the S protein of SARS-CoV-2 in a different manner. Nelfinavir mesylates was docked into the S2 subunit of SARS-CoV S protein (Fig. 7a). The same inhibitor was docked into to the N-terminal domain (NTD) region of the S1 subunit of SARS-CoV-2 and MERS-CoV S protein (Figs. 6a and 8a). 3-benzoylhosloppone with the highest binding affinity for SARS-CoV-2 S protein interacted via H-bond to THR547; Alkyl interaction to PHE541 and Pi-Alkyl interaction to PRO589 and LEU546. The region of interaction was between the CTD and SD1 region of S1 subunit of SARS-CoV-2 S protein. Cucurbitacin B was docked to the S2 subunit of SARS-CoV-2 S protein but interacted with different amino acid residue. The interaction of cucurbitacin B to the protein was via H-bond to ARG1091, ASN914, THR912, and GLN1113; Pi-Sigma bond to PHE1121 and Alkyl interaction to ILE1114 and GLY1124 (Fig. 6c). The same pattern of interaction was observed in both 7-Deacetoxy-7-oxogedunin and 3-friedelanone to the S2 subunit of SARS-CoV S protein. Both terpenoids interacted via a H-bond to ARG982 and GLY726 of the S2 subunit. While 7-deacetoxy-7-oxogedunin interacted with the upstream helix and central helix, 3-friedelanone interacted with the connecting region of the S2 subunit. A hydrophobic interaction via Pi-Alkyl and alkyl bonds was observed with the remaining amino acid residue (Table 2; Fig. 7b, c). 7-Deacetoxy-7-oxogedunin interacted via H-bond to the SER51 residue of N-terminal domain of the S1 subunit of MERS-CoV S protein. A Pi-Pi T-shaped interaction was formed between 7-deacetoxy-7-oxogedunin and PHE354; HIS670 of MERS-CoV S protein. Other hydrophobic interactions via Pi-Alkyl and Pi-Sigma bonds were observed to with the remaining amino acid residues (Table 4; Fig. 8a, b). 3-benzoylhosloppone interacted via: Pi-Sigma interaction to (PHE341) of NTD; Pi-Pi Stacking to (MET698) of SD2; Pi-Alkyl interaction to (LYS689) of SD2; and an Alkyl interaction to (LEU344 and ILE337) of NTD with the S1 subunit of MERS-CoV S protein (Fig. 8c). In summary, the binding of ligands to various proteins revealed eight terpenoid with remarkable binding affinities. Those with very good interactions with ACE2 and TMPRSS2 are 24-methylene cycloartenol; isoiguesterin; 11-hydroxy-2-(3,4-dihydroxybenzoyloxy) abieta-5,7,9(11),13-tetraene-12-one; and 11-hydroxy-2-(4-hydroxybenzoyloxy)-abieta-5,7,9(11),13-tetraene-12-one. Similarly, 3-benzoylhosloppone, and cucurbitacin B interacted well with SARS-CoV-2 spike protein, while 7-deacetoxy-7-oxogedunin and 3-friedelanone interacted well with SARS-CoV and MERS-CoV spike protein.

Visualization of interacting amino acid residues of human ACE2 with ligands in 3D (i) and 2D (ii) representation. Ligands in stick representation are presented in different colors. a Green: S1 (MLN-4760). b Read: 24-methylene cycloartenol. c Blue: isoiguesterin. Types of interactions are represented by green-dotted lines: H-bond interactions, light purple-dotted line: hydrophobic interactions (Pi-Alkyl, Alkyl, and pi-stacking) purple-dotted line: Pi-Pi T-shaped, yellow-dotted lines: Pi-sulphur interactions, pi-stacking interactions. Three-letter amino acids are in red color

Amino acid interactions of terpenoids in substrate binding cavity of human TMPRSS2. S Surface representation. Ligands in sticks representation are represented by colors. a Red: camostat. b Blue: 11-hydroxy-2-(3,4-dihydroxybenzoyloxy)abieta-5,7,9(11),13-tetraene-12-one. c Green: 11-hydroxy-2-(4-hydroxybenzoyloxy)-abieta-5,7,9(11),13-tetraene-12-one. Types of interactions are represented by green-dotted lines: H-bond interactions, light purple-dotted line: hydrophobic interactions (Pi-Alkyl, Alkyl, and pi-stacking) purple-dotted line: Pi-Pi T-shaped, yellow-dotted lines: Pi-sulphur interactions, pi-stacking interactions

Visualization of interacting amino acid residues of SARS-CoV-2 spike protein with ligands in 3D (i) and 2D (ii) representation. Ligands in stick representation are presented in different colors. a Green: nelfinavir mesylates (S3). b Red: 3-benzoylhosloppone. c Blue: cucurbitacin B. Types of interactions are represented by green-dotted lines: H-bond interactions, light purple-dotted line: hydrophobic interactions (Pi-Alkyl, Alkyl, and pi-stacking) purple-dotted line: Pi-Pi T-shaped, yellow-dotted lines: Pi-sulphur interactions, pi-stacking interactions

Visualization of interacting amino acid residues of SARS-CoV spike protein with ligands in 3D (i) and 2D (ii) representation. Ligands in stick representation are presented in different colors. a Green: nelfinavir mesylates (S3) b Red: 7-deacetoxy-7-oxogedunin. c Blue: 3-friedelanone.. Types of interactions are represented by green-dotted lines: H-bond interactions, light purple-dotted line: hydrophobic interactions (Pi-Alkyl, Alkyl, and pi-stacking) purple-dotted line: Pi-Pi T-shaped, yellow-dotted lines: Pi-sulphur interactions, pi-stacking interactions

Visualization of interacting amino acid residues of MERS-CoV spike protein with ligands in 3D (i) and 2D (ii) representation. Ligands in stick representation are presented in different colors. a Green: nelfinavir mesylates (S3). b Red: 7-deacetoxy-7-oxogedunin. c Blue: 3-benzoylhosloppone. Types of interactions are represented by green-dotted lines: H-bond interactions, light purple-dotted line: hydrophobic interactions (Pi-Alkyl, Alkyl, and pi-stacking) purple-dotted line: Pi-Pi T-shaped, yellow-dotted lines: Pi-sulphur interactions, pi-stacking interaction
Energy profile of best docked terpenoids to respective proteins
The overall energy profiles of terpenoid-protein complexes in the selected clusters with the best docked poses are shown in Figures S1– (supplementary data). Figure S1a–a (supplementary data) shows the breakdown of the binding energy of the selected cluster into different contributions. Gauss 1 (blue) and 2 (leaf green) bars represent the non-bonding interactions, red bar: repulsion, light blue bar: hydrophobic, purple bar: hydrogen bonds, light green bar: rotational forces, while the black bar represents total binding affinity which is a representative contribution of all bonding and non-bonding interactions between the terpenoids and the protein residues. The contributions of the various type of interaction as presented in graph (Figures S1a–a: supplementary data) shows that of the total binding energy of − 9.7 Kcal/mol exhibited by the binding of 24-methylene cycloartenol to the ACE2, − 2.1 and 1.8 Kcal/mol of hydrophobic and H-bond energies respectively was contributed, while the rest were contributed by non-bonding interaction mainly van der Waals, repulsive, and rotational forces. A H-bond, hydrophobic interaction, and repulsive energy of − 2.8 − 0.8, and + 2.3 Kcal/mol respectively was contributed to the total binding energy of − 10.0 Kcal/mol between T3 and TMPRSS2. Hydrophobic interaction affinities of − 2.1, − 0.6, and − 1.5 Kcal/mol, an H-bond energies of 0.3, − 0.6, and − 0.3 Kcal/mol were contributed to the total binding energy of the spike protein of SARS-CoV-2, SARS-CoV, and MERS-CoV with respective terpenoids. The rest of the energy was contributed by non-binding interactions.
Figures S1b–b (supplementary data) shows the overall energy profile of the ligand-receptor complex of the selected cluster, showing the individual energetic contributions for each atom in the ligand.
Molecular dynamics simulation
Four compounds including camostat, T3, 24-methylene cycloartenol, and 3-benzoylhosloppone were analyzed for their interactions with transmembrane protease serine 2 (TMPRSS2), and Angiotensin-converting enzyme 2 (ACE2) and SARS-CoV-2 Spike glycoprotein (S protein). Molecular dynamics simulation was done on each of the target protein-terpenoids complexes and the trajectories were analyzed. The radius of gyration (RoG), root mean square deviation (RMSD), root mean square fluctuation (RMSF), and surface accessible surface area (SASA) results were calculated for each trajectory. The RoG values give indication on the folding/unfolding of the protein. There was no observed difference between the RoG of TMPRSS2_camostat and TMPRSS2_T3 complexes (Fig. 9a). The TMPRSS2_cemostat, TMPRSS2_T3, and ACE2_ 24-methylene cycloartenol complexes show a steady fluctuation around mean values of 16.77 Å, 16.75 Å, 25.95 Å, while the RoG values of the S protein_3-benzoylhoslopponecomplex are the most fluctuating. The RMSD values show the deviation of each frame from the initial configuration (Fig. 9a, b). The average RMSD values from the plots of the TMPRSS2_camostat (2.13 Å) and TMPRSS2_T3 (2.14 Å) system were very close, while the ACE2-24_methylene cycloartenol and S protein_3-benzoylhosloppone complexes are around 3.6 Å and 16.78 Å, respectively (Figs. 10 and 11). The SASA plots indicate the rate of conformational changes in the protein based on its solvent accessibility. TMPRSS2_cemostat, TMPRSS2_T3, ACE2_24-methylene cycloartenol, and S protein 3-benzoylhosloppone complexes have average values of 11563 Å2, 11498 Å2, 29667 Å2, and 53680 Å2 (Fig. 10). The RMSF plots give information on the fluctuation of individual amino acids. All the four complex systems have spikes at the end of RMSF plots that indicates the motion of the terminals. The mean RMSF values for TMPRSS2_camostat and TMPRSS2_T3 are 0.68 and 0.73 Å (Fig. 12a), while the ACE2_24-methylenecycloartenol and S protein_(3-benzoylhosloppone) complexes were fluctuating around 1.29 Å and 7.36 Å, respectively (Fig. 12b). The spikes in the middle and the start of the RMSF of ACE2_(24-methylene cycloartenol) complex between amino acid 265 and amino acid 443 and spikes in S protein_(3-benzoylhosloppone) complex corresponds to the loops in the two protein respectively (Fig. 12).

The radius of gyration plots for a TMPRSS2_camostat and TMPRSS2_11-hydroxy-2-(3,4-dihydroxybenzoyloxy)abieta-5,7,9(11),13-tetraene-12-one and b ACE2_24-methylene cycloartenol and S protein-3-benzoylhosloppone complexes

The surface accessible surface area (SASA) plots for a TMPRSS2_camostat and TMPRSS2_ 11-hydroxy-2-(3,4-dihydroxybenzoyloxy)abieta-5,7,9(11),13-tetraene-12-one and b ACE2_24-methylene cycloartenol, SARS-CoV-2 S protein-3-benzoylhosloppone complexes

The root mean square deviation (RMSD) plots for a TMPRSS2_camostat and TMPRSS2_11-hydroxy-2-(3,4-dihydroxybenzoyloxy)abieta-5,7,9(11),13-tetraene-12-one and b ACE2_24-methylene cycloartenol and S protein-3-benzoylhosloppone complexes

The root mean square fluctuation plots for a TMPRSS2_(11-hydroxy-2-(3,4-dihydroxybenzoyloxy)abieta-5,7,9(11),13-tetraene-12-one) and TMPRSS2_camostat. b SARS-CoV-2 S protein_(3-benzoylhosloppone). c ACE2_(24-methylene cycloartenol) respectively
Clustering analysis of the MD simulation trajectory of complexes
Table S3 (Supplementary data) show the number of clusters, interaction number, and types for TMPRSS2_T3, TMPRSS2_cemostat, S protein_3-benzoylhosloppone, and ACE2_24-methylene cycloartenol, respectively. Hydrophobic, H-bond, and salt-bridges interactions were observed from PLIP webserver. Figure S4 (supplementary data) shows the first and last cluster representatives for the protein-terpenoids complexes and the mode of interaction in the enlarged part of the image. Images were generated using PyMol software V 2.2.2.
Molecular mechanics/generalized born surface area and decomposition analysis
MM/GBSA free energy decomposition analysis was employed to decompose the total binding free energies (ΔGbind) into terpenoid-residue pairs, which would provide more detailed information regarding the contribution of each residue for ligand binding. It is obvious that the residue spectrograms of the TMPRSS2 systems were similar, though with different intensity of interactions. The high binding free energy of reference inhibitor (camostat) to TMPRSS2 was predominately through its interaction with APS199 and ASP228. Other H-bonds (ALA190, ASP189, and ALA192) contributed immensely to the free energy. The top docked terpenoid (T3) had stronger binding affinities to the residues ARG41 of TMPRSS2 than camostat. Both spectra show fluctuations around ARG41. The results of the energetic calculations that is presented in Table 3 show that the two TMPRSS2 systems had close values for ∆Evdw, ∆GELE, and ∆GSA. The high ∆Gtotal of camostat_ TMPRSS2 as compared to the T3_ TMPRSS2 may have been contributed by the ∆Eele and ∆GGB. The decomposition plot for the ACE2 _24-methylene cycloartenol system and the SARS-CoV-2 S protein_3-benzoylhosloppone are in agreement with the results from the static docking analysis. The free binding energy of 24-methylene cycloartenol to ACE2 was majorly contributed by the H-bonds to SER167 and SER170 with the free energy contributions of which were greater than 1 kcal/mol. Other hydrogen bonds TRY497 and hydrophobic contacts to VAL491 were observed on the plot. The binding free energy of 3-benzoylhosloppone to SARS-Cov-2 S protein was majorly contributed by the H-bond and hydrophobic contact to THR547 other contributing residues includes LEU546, PHE565, VAL576, and ILE587 (Fig. 13).

Molecular mechanics/generalized born surface area (MM/GBSA) plot of binding free energy contribution per residue of a TMPRSS2_11-hydroxy-2-(3,4-dihydroxybenzoyloxy)abieta-5,7,9(11),13-tetraene-12-one. b TMPRSS2_camostat. c SARS-CoV-2 S protein_3-benzoylhosloppone. d ACE2 _24-methylene cycloartenol
Drug likeness and pharmacokinetic properties of selected terpenoids
The result generated from the Lipinski and ADMET filtering analyses are represented in Table 4 and Figure S5 (supplementary file). Four terpenoids T1, T3, T5, and T6 fulfilled the requirement for Lipinski analysis of the rule-of-five with corresponding favorable predicted ADMET parameters. The in silico predictive pharmacokinetic and ADMET properties from the filtering analyses suggested T1, T3, T5, and T6 with a high probability of absorption, subcellular distribution, and low toxicity. Though pharmacokinetic analysis indicated T1 (Table 4) to be less soluble while the ADME/tox analysis indicated high aqueous solubility, ability to pass the high human intestinal absorption, low acute oral toxicity with a good bioavailability score as exhibited by T3, T5, and T6 (Table 4).
Discussion
The prediction of drug–target interactions especially in new proteins is an essential stage in the drug discovery and development process [33]. Interference with several proteins that mediate viral attachment, membrane fusion, and cell entry of coronaviruses is an emerging therapeutic strategy for preventing COVID-19 infection [7, 20]. This principle was earlier demonstrated with HIV [13, 19] and SARS-CoV [2]. Earlier screening and prospecting of therapeutic phytocompounds have been reported for both SARS-CoV and MERS-CoV [42, 46, 50, 65]. Cell-based assays have shown the antiviral potentials of specific plant terpenoids against severe acute respiratory syndrome coronavirus (SARS- CoV) [65, 70]. This study was therefore undertaken to identify plant-derived terpenoids with inhibitory potentials against membrane-mediated SARS-CoV-2 entry proteins. Specifically, two triterpenes namely 24-methylene cycloartenol and isoiguesterin were reported to target ACE2 as well as the host-virus interface (S-protein-ACE2 receptor complex). These compounds interacted with adjacent residues in the conserved domain, apparently portraying its ability to bind and block interactions of hotspot 31 residues. The residues near lysine 31, and tyrosine 41, 82–84, and 353–357 in human ACE2 are important for the binding of S-protein of coronavirus [28]. The hotspots, 31 and 353, make salt bridge between Lys31 and Glu35, and the hotspot 353, comprising a salt bridge between Lys353 and Asp38, and are both buried in hydrophobic environment; therefore, interaction within this region is suggested to affect the binding of its substrate [69]. In a similar study in which five selected phytochemicals from Chinese and Indian herbs, though the individual compounds interacted differently with the active site of ACE2, they tend to distort the conformation that is necessary for its binding to the viral S protein [4]. The binding interactions of 24-methylene cycloartenol and isoiguesterin to the Site-2 binding site of ACE2 were similar to the pattern exhibited by some repurposed drugs such as delapril and lisinopril perindopril [24]. Abietane diterpenes, namely 11-hydroxy-2-(3,4-dihydroxybenzoyloxy) abieta-5,7,9(11), 13-tetraene-12-one (T3), and 11-hydroxy-2-(4-hydroxybenzoyloxy)-abieta-5,7,9(11), 13-tetraene-12-one (T4) showed the strongest interaction with TMPRSS2. In a similar binding pattern to camostat, these compounds were fitted into the S1-specificity pocket. They interacted with residue ALA190, ASP189, and GLN192 that are known to be part of the amino acid found at the basement of the pocket. ASP189 at the bottom of the pocket is known to determine the specificity of the S1 pocket for basic residues Arg and Lys at position P1 of the substrate [26]. The result showed that the hydroxybenzoyloxyl moiety of the terpenoids (T3 and T4) was responsible for at least 75% of the H-Bond with the protein. It was further observed that just as in the case of benzamidine (the native ligand) and camostat, the hydroxybenzoyloxyl moiety of the two terpenoids points with its hydroxyl group towards the carboxylate group of ASP189 forming strong H-bonds with ASP189 and other residue in the pocket. For camostat, the phenylquanidine moiety pointed into the hydrophobic pocket with the negatively charged ASP189 at its bottom. Unlike the H-bond formed between the amidino nitrogen of the phenylquanidine and benzamidine, in T3 and T4 the H-Bonds were formed mainly with the hydroxyl and carboxylate group. A striking similarity observed was that the ester bond that linked both the phenylquanidine moiety of camostat and the hydroxybenzoyloxyl moiety of T3 and T4 to the remaining structural unit of the compounds formed strong H-Bonds to the same residue SER195. The phenyl group of the hydroxybenzoyloxy moiety of T3 and T4 further interacted with hydrophobic interactions to CYS119 and CYS219 just as the peptide planes of the bonds between TRP215–GLY216 and CYS191–GLN192 sandwich the phenyl ring of benzamidine [16, 26]. The additional hydrophobic interaction by T3 and T4 may have been responsible for the exhibited higher binding affinities than camostat and benzamidine. Furthermore, while the hydroxybenzoyloxy moiety was directed towards the hydrophobic cleft created by ASP189, the abietane agylcon interacted with the imidazol ring of HIS57 of the S2 pocket that is found next to the S1 pocket and ARG41 (in the case of T4) which are outside the hydrophobic cleft. A similar interaction as the later was observed with camostat. The strong similarity in the binding pattern and even a far strong binding affinity than camostat and benzamidine indicates that T3, T4, and other abietane diterpenes especially those with hydroxybenzoyloxyl moiety attached to the abietane aglycon are potential inhibitors of TMPRSS2, thus preventing some coronaviruses from entering host [26]. Some natural compounds were found to interact with the protease furan of TMPRSS2, and these compounds exhibited different binding modes in the active site [52, 62]. It is known that, like SARS-CoV, SARS-CoV-2 S protein recognizes and binds to host-cell receptor angiotensin-converting enzyme 2 (ACE2) using a transmembrane protease serine 2 (TMPRSS2) which activates the S protein to facilitate viral fusion and entry into cells [68]. It is important to note that serine protease inhibitors like camostat mesylate, which blocks the activity of TMPRSS2 [77], has been approved in Japan for human use. Related compounds with antiviral activity potentiates as anti-SARS-CoV-2 agent [71]. Also, some abietane terpenoids have been identified to exhibit in vitro anti-SARS-CoV activity [65]. This corroborates the result of our study that shows that abietane diterpenes exhibits a wide spectrum and multiplicity of protein binding and may thereby specifically execute a complete blockage of viral entry. With regard to coronavirus S-proteins, 3-benzoylhosloppone and cucurbitacin B were the two terpenoids of utmost interest. While 3-benzoylhosloppone interacted with amino acid residue of the RBD and SD1 region of the S1 subunit, cucurbitacin B was docked into the S2 subunit of SARS-CoV-2 S protein. The former subunit is responsible for receptor recognition while the later mediates the fusion of viral membrane and the host cellular membrane [76]. Some phytochemicals known to interact with the RBD region and other binding site of the SARS-CoV-2 S protein have been reported to disrupt the binding of the S protein to the ACE2 protein [4, 45]. These terpenoids may prevent interaction of spike protein with its host cell receptor, thereby preventing entry of virus into host cell. 3-benzoylhosloppone has been reported for its antimalarial property while cucurbitacin B is an anticancer agent [1, 15].
Molecular dynamics (MD) simulations was performed after docking analysis to assess the physical transitions of atoms to effectively adopt the structure-to-function relevance of top docked terpenoids-target proteins and to further understand the dynamic behavior of the top docked terpenoids in the binding site of the various conformations of the target protein complexes in a dynamic environment [75]. The stability and structural/conformational fluctuations that occurred in the target proteins-terpenoids systems were monitored by clustering analysis of the MDS trajectory files. The RMSD is a plausible measure of protein stability. RMSD data shows how much each frame is deviated from the initial conformation of the reference structure as a function of time [11]. The comparison of the RMSD plots for the camostat_TMPRSS2 and T4_TMPRSS2 systems shows that the binding of T3 did not cause any structure deformation in TMPRSS2 as the binding of camostat. RMSF indicates the flexibility of different regions of a protein and the amino acid residue along the trajectory, which can be related to crystallographic B factors [11]. Though a lower amount of fluctuation occurred at with the interacting residues, it has been established that greater amounts of structural fluctuations usually occur in regions known to be involved in ligand binding and catalysis, notably the catalytic loop regions [14]. The RoG and SASA were assessed to evaluate the structural compactness and the accessibility of solvent to the proteins. A stably folded protein maintains a reasonably steady RoG over the simulation time. The stability of the complex is affected by loss of compactness through the introduction of weak intermolecular bonds [51].The RoG and SASA plots of all the systems did not show fluctuation that indicates deformation of the structural integrity of the proteins. The analyses of the thermodynamic parameters of the systems show that the top docked terpenoid complexed with respective proteins targets were stable and could be therefore subjected to experimental processes in further studies. At a quantitative level, simulation-based methods provide substantially more accurate estimates of ligand binding affinities (free energies) [43]. These results are calculated based on the total binding free energy of the complex. In these calculations, the binding free energy (∆Gbind) measures the affinity of a ligand to its target protein. The free energy difference between the ligand-bound state (complex) and the corresponding unbound states of proteins and ligands are also employed in the calculations. Thus, the ∆Gbind calculations are important to gain in-depth knowledge about the binding modes of the hits in drug design [25]. The result from the MMPBSA calculation further corroborated the docking studies. The same amino acid residues were involved in the interactions with the top docked terpenoids in the static and dynamic states. From the Lipinski, pharmacokinetic, and ADMET filtering analyses, we identified four druggable and non-toxic, natural terpenoids that exhibited strong binding tendency to the various protein targets that mediates coronavirus-host cell entry. The result from the predicted filtering analyses of the four compounds showed parameters that suggest a favorable in silico ADMET and pharmacokinetic properties. The terpenoids expressed high probability of human intestinal absorption. They were also non-substrate to the permeability-glycoprotein (P-gp) [29], expressed capability to cross the blood brain barrier (BBB). SARS-CoV-2 has been reported to infect the brain, thus indicating its ability to cross the blood brain barrier (BBB) [73]. Therefore, compounds that can cross the BBB will be beneficial in the overal all viral clearance. The four terpenoids did not show indication of mutagenicity in silico, thereby they may not cause genetic mutations. The compounds did not display inhibitory potential for the various cytochrome P450, thus may not adversely affect phase I drug metabolism in the liver. These terpenoids are therefore considered as potential drug candidates.
Conclusions
A virtual screening approach was successfully applied to identify plant-derived terpenoids as potential inhibitor of coronavirus cells entry proteins. Two pentacyclic terpenoids (4-methylene cycloartenol and isoiguesterin) interacted strongly with the binding sites residues that are known to interfere with the activity of ACE2. The abietane diterpene especially: 11-hydroxy-2-(3,4-dihydroxybenzoyloxy) abieta-5,7,9 (11), 13-tetraene-12-one (T3), and 11-hydroxy-2-(4-hydroxybenzoyloxy)-abieta-5,7,9(11), 13-tetraene-12-one (T4) exhibited a similar binding pattern to the S1-specificity pocket of TMPRSS2 as camostat (reference inhibitor). They also showed wide spectrum and multiplicity of entry protein binding. The terpenoids binding conformations in the complexes were stable in a simulated dynamic environment. The MM-GBSA binding free energy calculations corroborated the static docking analysis. Since the identified lead terpenoids showed drug-likeness and low toxicity as indicated by the in silico pharmacokinetically relevant molecular descriptors, they are postulated as potential inhibitors that can be considered for further in vitro and in vivo studies towards developing entry inhibitors against the ongoing coronavirus pandemic.
Availability of data and materials
The authors confirm that the data supporting the findings of this study are available within the article [and/or] its supplementary materials.
Abbreviations
- COVID-19:
-
Coronavirus disease-19
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus 2
- SARS-CoV:
-
Severe acute respiratory syndrome coronavirus
- MERS-CoV:
-
Middle East respiratory syndrome coronavirus
- TMPRSS2:
-
Transmembrane protease serine 2
- ACE2:
-
Angiotensin-Converting Enzyme 2
- ADMET:
-
Absorption, distribution, metabolism, excretion, and toxicity
- MM-GBSA:
-
Mechanics–generalized born surface area
- RMSD:
-
Root mean square deviation
- RMSF:
-
Root mean square fluctuation
- SASA:
-
Surface accessible surface area
- RoG:
-
Radius of gyration
- PBC:
-
Periodic boundary conditions
References
Achenbach H, Waibel R, Nkunya MH, Weenen H (1992) Antimalarial compounds from Hoslundia opposita. Phytochemistry 31:3781–3784
Adedeji A, Severson W, Jonsson C, Singh K, Weiss S, Sarafianos S (2013) Novel inhibitors of SARS-CoV entry acting by three distinct mechanisms. J Virol 87(14):87–8028. https://doi.org/10.1128/JVI.00998-13
Bagla V, McGaw L, Eloff J (2012) The antiviral activity of six South African plants traditionally used against infections in ethnoveterinary medicine. Vet Microbiol 155:198–206. https://doi.org/10.1016/j.vetmic.2011.09.015
Basu A, Sarkar A, Maulik U (2020) Molecular docking study of potential phytochemicals and their effects on the complex of SARS-CoV2 spike protein and human ACE2. Sci Rep 10:1–15
Beuscher N, Bodinet C, Neumann-Haefelin D, Marston A, Hostettmann K (1994) Antiviral activity of African medicinal plants. J Ethnopharmacol 42(2):101–109. https://doi.org/10.1016/0378-8741(94)90103-1
Brooks BR, Brooks CL III, Mackerell AD Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M (2009) CHARMM: the biomolecular simulation program. J Comput Chem 30(10):1545–1614. https://doi.org/10.1002/jcc.21287
Chen H, Du Q (2020) Potential natural compounds for preventing SARS-CoV-2 (2019-nCoV) infection
Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, Qiu Y, Wang J, Liu Y, Wei Y, Xia J’, Yu T, Zhang X, Zhang L (2020a) Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet (London, England) 395:507–513. https://doi.org/10.1016/s0140-6736(20)30211-7
Chen YW, Yiu C-P, Wong K-Y (2020b) Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CLpro) structure: virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Research 9:129. https://doi.org/10.12688/f1000research.22457.1
Cheng F, Li W, Zhou Y, Shen J, Wu Z, Liu G, Lee PW, Tang Y (2012) admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model 52:3099–3105. https://doi.org/10.1021/ci300367a
Cheng X, Ivanov I (2012) Molecular dynamics. Comput Toxicol 929:243–285
Coutard B, Valle C, de Lamballerie X, Canard B, Seidah NG, Decroly E (2020) The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Res 176:104742. https://doi.org/10.1016/j.antiviral.2020.104742
Derdeyn CA, Decker JM, Sfakianos JN, Wu X, O'Brien WA, Ratner L, Kappes JC, Shaw GM, Hunter E (2000) Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J Virol 74:8358–8367. https://doi.org/10.1128/jvi.74.18.8358-8367.2000
Dong Y-w, Liao M-l, X-l M, Somero GN (2018) Structural flexibility and protein adaptation to temperature: Molecular dynamics analysis of malate dehydrogenases of marine molluscs. Proc Natl Acad Sci 115:1274–1279
Garg S, Kaul SC, Wadhwa R (2018) Cucurbitacin B and cancer intervention: chemistry, biology and mechanisms. Int J Oncol 52:19–37
Gyebi GA, Adegunloye AP, Ibrahim IM, Ogunyemi OM, Afolabi SO, Ogunro OB (2020a) Prevention of SARS-CoV-2 cell entry: insight from in silico interaction of drug-like alkaloids with spike glycoprotein, human ACE2, and TMPRSS2. J Biomol Struct Dyn p 1–25. https://doi.org/10.1080/07391102.2020.1835726
Gyebi GA, Ogunro OB, Adegunloye AP, Ogunyemi OM, Afolabi SO (2020b) Potential inhibitors of coronavirus 3-chymotrypsin-like protease (3CLpro): an in silico screening of alkaloids and terpenoids from African medicinal plants. J Biomol Struct Dyn 39:3396-3408. https://doi.org/10.1080/07391102.2020.1764868
Gyebi GA, Ogunyemi OM, Ibrahim IM, Afolabi SO, Adebayo JO (2021) Dual targeting of cytokine storm and viral replication in COVID-19 by plant-derived steroidal pregnanes: an in silico perspective. Comput Biol Med 134:104406
Hartt JK, Liang T, Sahagun-Ruiz A, Wang JM, Gao JL, Murphy PM (2000) The HIV-1 cell entry inhibitor T-20 potently chemoattracts neutrophils by specifically activating the N-formylpeptide receptor. Biochem Biophys Res Commun 272:699–704. https://doi.org/10.1006/bbrc.2000.2846
Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu NH, Nitsche A, Müller MA, Drosten C, Pöhlmann S (2020) SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor cell. https://doi.org/10.1016/j.cell.2020.02.052
Hui DS, I Azhar E, Madani TA, Ntoumi F, Kock R, Dar O, Ippolito G, Mchugh TD, Memish ZA, Drosten C, Zumla A, Petersen E (2020) The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health - The latest 2019 novel coronavirus outbreak in Wuhan, China. Int J Infect Dis 91:264–266. https://doi.org/10.1016/j.ijid.2020.01.009
Humphrey W, Dalke A, Schulten K (1996a) VMD: visual molecular dynamics. J Mol Graph 14:33–38
Jaeger R, Cuny E (2016) Terpenoids with special pharmacological significance: a review. Nat Prod Commun 11:1934578X1601100946. https://doi.org/10.1177/1934578X1601100946
Khelfaoui H, Harkati D, Saleh BA (2020) Molecular docking, molecular dynamics simulations and reactivity, studies on approved drugs library targeting ACE2 and SARS-CoV-2 binding with ACE2. J Biomol Struct Dyn p 1–17. https://doi.org/10.1080/07391102.2020.1803967
Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE (2000) Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res 33(12):889–897. https://doi.org/10.1021/ar000033j
Kyrieleis OJ, Huber R, Ong E, Oehler R, Hunter M, Madison EL, Jacob U (2007) Crystal structure of the catalytic domain of DESC1, a new member of the type II transmembrane serine proteinase family. FEBS J 274(8):2148–2160. https://doi.org/10.1111/j.1742-4658.2007.05756.x
Lee J, Cheng X, Swails JM, Yeom MS, Eastman PK, Lemkul JA, Wei S, Buckner J, Jeong JC, Qi Y, Jo S, Pande VS, Case DA, Brooks CL III, MacKerell AD Jr, Klauda JB, Im W (2016) CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J Chemical Theory Comput 12(1):405–413. https://doi.org/10.1021/acs.jctc.5b00935
Li W et al (2005) Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J 24:1634–1643
Lin JH, Yamazaki M (2003) Role of P-glycoprotein in pharmacokinetics. Clin Pharmacokinet 42(1):59–98. https://doi.org/10.2165/00003088-200342010-00003
Lipinski CA (2000) Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods 44:235–249. https://doi.org/10.1016/s1056-8719(00)00107-6
Liu C, Zhou Q, Li Y, Garner LV, Watkins SP, Carter LJ, Smoot J, Gregg AC, Daniels AD, Jervey S, Albaiu D (2020) Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases. ACS Central Sci 6(3):315–331. https://doi.org/10.1021/acscentsci.0c00272
Mahmud SH et al (2021a) Bioinformatics and system biology approach to identify the influences of SARS-CoV-2 infections to idiopathic pulmonary fibrosis and chronic obstructive pulmonary disease patients. Brief Bioinform. bbab115, https://doi.org/10.1093/bib/bbab115
Mahmud SH, Chen W, Liu Y, Awal MA, Ahmed K, Rahman MH, Moni MA (2021b) PreDTIs: prediction of drug–target interactions based on multiple feature information using gradient boosting framework with data balancing and feature selection techniques. Brief Bioinform. bbab046, https://doi.org/10.1093/bib/bbab046
Mark P, Nilsson L (2001) Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. Chem A Eur J 105(43):9954–9960. https://doi.org/10.1021/jp003020w
Miller BR III, McGee TD Jr, Swails JM, Homeyer N, Gohlke H, Roitberg AE (2012) MMPBSA. py: an efficient program for end-state free energy calculations. J Chemical Theory Comput 8(9):3314–3321. https://doi.org/10.1021/ct300418h
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem 30:2785–2791
Ndhlala A, Amoo S, Ncube B, Moyo M, Nair J, van Staden J (2013) Antibacterial, antifungal, and antiviral activities of African Medicinal Plants, pp 621–651. https://doi.org/10.1016/B978-0-12-405927-6.00016-3
Nickel J, Gohlke BO, Erehman J, Banerjee P, Rong WW, Goede A, Dunkel M, Preissner R (2014) SuperPred: update on drug classification and target prediction. Nucleic Acids Res 42:W26–W31. https://doi.org/10.1093/nar/gku477
O'Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR (2011) Open Babel: An open chemical toolbox. J Chem 3:33. https://doi.org/10.1186/1758-2946-3-33
Ogunyemi OM, Gyebi GA, Elfiky AA, Afolabi SO, Ogunro OB, Adegunloye AP, Ibrahim IM (2020) Alkaloids and flavonoids from African phytochemicals as potential inhibitors of SARS-Cov-2 RNA-dependent RNA polymerase: an in silico perspective. Antiviral Chem Chemother 28:2040206620984076
Ojo O, Oluyege J, Famurewa O (2009) Antiviral properties of two Nigerian plants. Afr J Plant Sci 3:157–159
Park JY, Kim JH, Kim YM, Jeong HJ, Kim DW, Park KH, Kwon HJ, Park SJ, Lee WS, Ryu YB (2012) Tanshinones as selective and slow-binding inhibitors for SARS-CoV cysteine proteases. Bioorg Med Chem 20:5928–5935. https://doi.org/10.1016/j.bmc.2012.07.038
Perez A, Morrone JA, Simmerling C, Dill KA (2016) Advances in free-energy-based simulations of protein folding and ligand binding. Curr Opin Struct Biol 36:25–31. https://doi.org/10.1016/j.sbi.2015.12.002
Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K (2005) Scalable molecular dynamics with NAMD. J Comput Chem 26(16):1781–1802. https://doi.org/10.1002/jcc.20289
Pushkaran AC, Melge AR, Puthiyedath R, Mohan CG (2021) A phytochemical-based medication search for the SARS-CoV-2 infection by molecular docking models towards spike glycoproteins and main proteases. RSC Adv 11:12003–12014
Ryu YB, Park SJ, Kim YM, Lee JY, Seo WD, Chang JS, Park KH, Rho MC, Lee WS (2010) SARS-CoV 3CLpro inhibitory effects of quinone-methide triterpenes from Tripterygium regelii. Bioorg Med Chem Lett 20:1873–1876. https://doi.org/10.1016/j.bmcl.2010.01.152
Salentin S, Schreiber S, Haupt VJ, Adasme MF, Schroeder M (2015) PLIP: fully automated protein–ligand interaction profiler. Nucleic Acids Res 43(W1):W443–W447. https://doi.org/10.1093/nar/gkv315
Sanchez-Linares I, Perez-Sanchez H, Cecilia JM, Garcia JM (2012) High-throughput parallel blind virtual screening using BINDSURF. BMC Bioinform 13(Suppl 14):S13. https://doi.org/10.1186/1471-2105-13-s14-s13
Shang J et al. (2020) Structural basis for receptor recognition by the novel coronavirus from Wuhan
Shen L, Niu J, Wang C, Huang B, Wang W, Zhu N, Deng Y, Wang H, Ye F, Cen S, Tan W (2019) High-throughput screening and identification of potent broad-spectrum inhibitors of coronaviruses. J Virol 93:e00023-19. https://doi.org/10.1128/jvi.00023-19
Sinha S, Wang SM (2020) Classification of VUS and unclassified variants in BRCA1 BRCT repeats by molecular dynamics simulation. Comput Struct Biotechnol J 18:723–736
Sonawane K et al. (2020) Homology modeling and docking studies of TMPRSS2 with experimentally known inhibitors camostat mesylate, nafamostat and bromhexine hydrochloride to control SARS-Coronavirus-2
Srinivasan S, Cui H, Gao Z, Liu M, Lu S, Mkandawire W, Narykov O, Sun M, Korkin D (2020) Structural genomics of SARS-CoV-2 indicates evolutionary conserved functional regions of viral proteins. Viruses 12:360. https://doi.org/10.3390/v12040360
Suomivuori C-M et al (2020) Molecular mechanism of biased signaling in a prototypical G protein–coupled receptor. Science (New York, NY) 367:881–887
Taniguchi Y, Nishikawa H, Maeda N, Terada Y (2020) Breathlessness, pleural effusions, fibromas, and Meigs syndrome: look beyond the chest and don't delay! Lancet 395:e32. https://doi.org/10.1016/S0140-6736(20)30111-2
Taz TA, Ahmed K, Paul BK, Al-Zahrani FA, Mahmud SH, Moni MA (2021a) Identification of biomarkers and pathways for the SARS-CoV-2 infections that make complexities in pulmonary arterial hypertension patients. Brief Bioinform 22:1451–1465
Taz TA, Ahmed K, Paul BK, Kawsar M, Aktar N, Mahmud SH, Moni MA (2021b) Network-based identification genetic effect of SARS-CoV-2 infections to Idiopathic pulmonary fibrosis (IPF) patients. Brief Bioinform 22:1254–1266
Towler P, Staker B, Prasad SG, Menon S, Tang J, Parsons T, Ryan D, Fisher M, Williams D, Dales NA, Patane MA, Pantoliano MW (2004) ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J Biol Chem 279:17996–18007. https://doi.org/10.1074/jbc.M311191200
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31:455–461. https://doi.org/10.1002/jcc.21334
Tubiana T, Carvaillo J-C, Boulard Y, Bressanelli S (2018) TTClust: a versatile molecular simulation trajectory clustering program with graphical summaries. J Chem Inf Model 58(11):2178–2182. https://doi.org/10.1021/acs.jcim.8b00512
Tung N, Kwon H-J, Kim J-H, Ra J, Ding Y, Kim J, Kim YH (2010) Antiinfluenza diarylheptanoids from the bark of Alnus japonica. Bioorg Med Chem Lett 20(3):1000–1003. https://doi.org/10.1016/j.bmcl.2009.12.057
Vardhan S, Sahoo SK (2021) Virtual screening by targeting proteolytic sites of furin and TMPRSS2 to propose potential compounds obstructing the entry of SARS-CoV-2 virus into human host cells. J Tradit Complement Med. https://doi.org/10.1016/j.jtcme.2021.04.001
Wan Y, Shang J, Graham R, Baric RS, Li F (2020) Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J Virol 94(7):e00127–e00120. https://doi.org/10.1128/JVI.00127-20
Wang Q et al (2020) Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 181:894–904. e899
Wen CC, Kuo YH, Jan JT, Liang PH, Wang SY, Liu HG, Lee CK, Chang ST, Kuo CJ, Lee SS, Hou CC, Hsiao PW, Chien SC, Shyur LF, Yang NS (2007) Specific plant terpenoids and lignoids possess potent antiviral activities against severe acute respiratory syndrome coronavirus. J Med Chem 50:4087–4095. https://doi.org/10.1021/jm070295s
WHO (2020) Report of the WHO-China joint mission on coronavirus disease 2019 (COVID-19) Geneva
Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, Graham BS, McLellan JS (2020) Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science (New York, NY) 367:eabb2507. https://doi.org/10.1126/science.abb2507
Wu C, Liu Y, Yang Y, Zhang P, Zhong W, Wang Y, Wang Q, Xu Y, Li M, Li X, Zheng M, Chen L, Li H (2020) Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharmaceut Sinica B 10(5):766–788. https://doi.org/10.1016/j.apsb.2020.02.008
Wu K, Li W, Peng G, Li F (2009) Crystal structure of NL63 respiratory coronavirus receptor-binding domain complexed with its human receptor. Proc Natl Acad Sci 106:19970–19974
Xiao S, Tian Z, Wang Y, Si L, Zhang L, Zhou D (2018) Recent progress in the antiviral activity and mechanism study of pentacyclic triterpenoids and their derivatives. Med Res Rev 38(3):951–976. https://doi.org/10.1002/med.21484
Yamamoto M, Matsuyama S, Li X, Takeda M, Kawaguchi Y, Inoue J-i, Matsuda Z (2016) Identification of nafamostat as a potent inhibitor of Middle East respiratory syndrome coronavirus S protein-mediated membrane fusion using the split-protein-based cell-cell fusion assay. Antimicrob Agents Chemother 60(11):6532–6539. https://doi.org/10.1128/AAC.01043-16
Yuan Y, Cao D, Zhang Y, Ma J, Qi J, Wang Q, Lu G, Wu Y, Yan J, Shi Y, Zhang X, Gao GF (2017) Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat Commun 8:15092. https://doi.org/10.1038/ncomms15092
Zanin L, Saraceno G, Panciani PP, Renisi G, Signorini L, Migliorati K, Fontanella MM (2020) SARS-CoV-2 can induce brain and spine demyelinating lesions. Acta Neurochir 162(7):1491–1494. https://doi.org/10.1007/s00701-020-04374-x
Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS (2020) Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med 46(4):586–590. https://doi.org/10.1007/s00134-020-05985-9
Zhao H, Caflisch A (2015) Molecular dynamics in drug design. Eur J Med Chem 91:4–14. https://doi.org/10.1016/j.ejmech.2014.08.004
Zhou P et al (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579:270–273
Zhou Y, Vedantham P, Lu K, Agudelo J, Carrion R Jr, Nunneley JW, Barnard D, Pöhlmann S, McKerrow JH, Renslo AR, Simmons G (2015) Protease inhibitors targeting coronavirus and filovirus entry. Antiviral Res 116:76–84. https://doi.org/10.1016/j.antiviral.2015.01.011
Acknowledgements
The authors appreciate the members of the BioNet-AP: Bioinformatics Network on African Phytomedicine for COVID-19 research. MD simulation and MM-GBSA calculations are done on the Bibliotheca Alexandrina HPC facility, Alexandria, Egypt.
Funding
The research did not receive any funding or grants.
Author information
Authors and Affiliations
Contributions
GAG: conceived and designed the analysis performed molecular docking. OMO: wrote manuscript. IMI: performed molecular simulations. OBO: editing and review of manuscript. APA: editing and review of manuscript. SOA: editing and review of manuscript. All authors have read and approved the final manuscript
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1
. Binding energies of bioactive terpenoids from African plants with higher affinity to human ACE2 and TMPRSS2, and SARS-Cov-2 S protein. Table S2. AutoDock scores (binding energies) of standard drugs and top 20 bioactive terpenoids with human Angiotensin-Converting Enzyme 2 (ACE2), Transmembrane Protease Serine 2 (TMPRSS2), and ACE2-Spike Receptor Binding Domain complex (ACE2-RBD). Table S3. AutoDock scores (binding energies) of standard drug and bioactive terpenoids from selected African phytochemicals to the spike protein of Coronaviruses. Table S4. Shows the number of clusters produced from TTClust, its representative frame for each of the protein-ligand complexes, and the interactions between the ligand and the protein from PLIP webserver for that frame. Figure S1. Energy profile of 24-methylene cycloartenol binding groups in human ACE2: (a) Energetic contribution to the Binding energy (d) Energetic contributions for each atom in the ligand. Number of poses in selected cluster: 68, best pose: 116 and binding site coordinate: 39.14, 35.33, and 12.71. Figure S2. Energy profile of T3 binding groups in human TMPRSS2: (a) Energetic contribution to the Binding energy (d) Energetic contributions for each atom in the ligand. Number of poses in selected cluster: 87, best pose: 40 and binding site coordinate: -2.96, 26.97, and 23.55. Figure S3. Energy profile of 3- benzoylhosloppone binding groups in SARS-Cov-2 S protein (a) Energetic contribution to the Binding (b) Energetic contributions for each atom in the ligand. Number of poses in selected cluster: 49, best pose: 571 and binding site coordinate: 214.85, 246.53, and 212.68. Figure S4. The representative structure for each cluster in cartoon representation, ligands in sticks representation and the types of interactions. Gray-dotted line: hydrophobic interactions, blue lines: H-bond interactions, yellow-dotted lines: salt-bridges interactions, and green-dotted lines: pi-stacking interactions. Single-letter amino acids are in red color. Figure S5. Summary of phamacokinetic properties of top binding terpenoids from African plants (a) T1: 24-methylene cycloartenol; (b) T3:11-Hydroxy-2 - (3,4-dihydroxybenzoyloxy) abieta -5,7,9(11),13-tetraene-12-one: (c) T5: 3- Benzoylhosloppone and (d) T6: Cucurbitacin B to the ACE2, TMPRSS2 and S protein of SARS-Cov-2.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gyebi, G.A., Ogunyemi, O.M., Ibrahim, I.M. et al. SARS-CoV-2 host cell entry: an in silico investigation of potential inhibitory roles of terpenoids. J Genet Eng Biotechnol 19, 113 (2021). https://doi.org/10.1186/s43141-021-00209-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43141-021-00209-z