
Abstract
Background
Accessing COVID-19 vaccines is a challenge despite successful clinical trials. This burdens the COVID-19 treatment gap, thereby requiring accelerated discovery of anti-SARS-CoV-2 agents. This study explored the potential of anti-HIV reverse transcriptase (RT) phytochemicals as inhibitors of SARS-CoV-2 non-structural proteins (nsps) by targeting in silico key sites in the structures of SARS-CoV-2 nsps. One hundred four anti-HIV phytochemicals were subjected to molecular docking with nsp3, 5, 10, 12, 13, 15, and 16. Top compounds in complex with the nsps were investigated further through molecular dynamics. The drug-likeness and ADME (absorption, distribution, metabolism, and excretion) properties of the top compounds were also predicted using SwissADME. Their toxicity was likewise determined using OSIRIS Property Explorer.
Results
Among the top-scoring compounds, the polyphenolic functionalized natural products comprised of biflavones 1, 4, 11, 13, 14, 15; ellagitannin 9; and bisisoquinoline alkaloid 19 were multi-targeting and exhibited strongest binding affinities to at least two nsps (binding energy = − 7.7 to − 10.8 kcal/mol). The top ligands were stable in complex with their target nsps as determined by molecular dynamics. Several top-binding compounds were computationally druggable, showed good gastrointestinal absorptive property, and were also predicted to be non-toxic.
Conclusions
Twenty anti-HIV RT phytochemicals showed multi-targeting inhibitory potential against SARS-CoV-2 non-structural proteins 3, 5, 10, 12, 13, 15, and 16. Our results highlight the importance of polyhydroxylated aromatic substructures for effective attachment in the binding/catalytic sites of nsps involved in post-translational mechanism pathways. As such with the nsps playing vital roles in viral pathogenesis, our findings provide inspiration for the design and discovery of novel anti-COVID-19 drug prototypes.
Similar content being viewed by others
Background
The rapid spread of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) marks itself as one of the deadliest viruses in recent history due to its high mortality and morbidity rates [1, 2]. As of May 2021, the World Health Organization recorded over one hundred sixty-seven million cases worldwide with 3.4 million deaths [3]. Continuous efforts are being carried out to unravel the pathophysiology of the virus, paving the way to the discovery and development of efficacious vaccines and anti-SARS-CoV-2 drugs. While the world continues to make strides in vaccine development and rollout, drug-based treatments are still needed to cure the growing number of COVID-19-afflicted individuals. Thus, developing effective therapeutic agents against SARS-CoV-2 remains a global health need.
The discovery of antiviral chemotherapeutic prototypes requires accurate identification of drug targets. Among which, the SARS-CoV-2 non-structural proteins (nsps) are among the highly favored targets because of their role in viral replication, post-translational mechanisms, and host immunity evasion that influence SARS-CoV-2 virulence and pathogenesis [4]. The repurposing of bioactive natural products is one of the key strategies available for screening potential SARS-CoV-2 nsps inhibitors. To date, plant-based medicines as treatment for SARS-CoV-2 infection have not been reported.
Plant-derived natural products are established biotechnological-derived substances that exhibit a wide range of biological activity including antagonistic properties against human immunodeficiency virus (HIV) and coronaviruses such as Middle East respiratory syndrome coronavirus (MERS-CoV) and SARS-CoV [5,6,7,8,9]. Relevant to this study, polyphenolic natural products such as flavonoids and tannins (Fig. 1) are well-recognized to confer broad-spectrum antiviral activities in addition to possessing anti-inflammatory, anti-tumor, antioxidants, immune, and prebiotic properties [10]. Recent studies in anti-COVID-19 drug discovery have highlighted the potential of polyphenolic compounds through in silico-guided investigations against protein targets in SARS-CoV-2 involved in infective mechanisms, i.e., inhibition of spike (S) protein, angiotensin-converting enzyme 2 (ACE-2) receptor, papain-like protease (PLpro), 3-chymotrypsin-like cysteine protease (3CLpro), and RNA-dependent-RNA-polymerase (RdRp) [10,11,12,13]. In addition, computational studies describing the potential of other classes of natural products as SARS-CoV-2 3CLpro, PLpro, and RdRp protein inhibitors have been reported [5, 14,15,16,17,18,19].

Anti-HIV RT phytochemicals with strong binding affinities to at least one of the target nsps
Considering the similarity between SARS-CoV-2 and HIV, we repurposed previously reported anti-HIV reverse transcriptase (RT) secondary compounds using in silico simulations in this study. SARS-CoV-2 and HIV are single-stranded RNA viruses that utilize RNA-dependent polymerases and code precursor polyproteins vital for their respective pathogenesis. In this paper, we disclose computational interrogation of 104 known anti-HIV RT phytochemicals against seven target proteins, namely nsp3 (PLpro), nsp5 (3CLpro), nsp12 (RdRp), nsp13 (helicase), nsp15 (endoribonuclease), and the nsp16-nsp10 complex (S-adenosylmethionine complex). The thermodynamic stability and the pharmacokinetic characteristics of the top-ranked compounds are also reported.
Methods
Target enzyme preparation
Seven target enzymes with important functions in SARS-CoV-2 infectivity were selected and obtained from the Protein Data Bank (PDB): 3CLpro (PDB ID: 6LU7), PLpro (PDB ID: 6W9C), RdRp (PDB ID: 6M71), helicase (6JYT), nsp16-nsp10 complex (6W4H), and nsp15 (6VWW). These proteins in three-dimensional structures were added to UCSF Chimera 1.14 platform as PDB files [20]. All proteins belong to SARS-CoV-2 except for helicase due to unavailability of nsp13. Thus, helicase model from SARS-CoV-1 which shares 99.8% sequence identity and 100% sequence similarity with that of SARS-CoV-2 was used [21]. Coronavirus helicase domains are distinct compared to other (+)-sense RNA virus domains due to the presence of linkage in a single protein to a binuclear zinc-binding domain at the N-terminus. This domain is composed of 12 conserved cysteine-histidine residues and is a good target in antiviral drug discovery [22,23,24,25].
Ligand selection and preparation
A total of 104 plant secondary metabolites (Supplementary Figure 1; Supplementary Table 1) previously reported to inhibit HIV RT [25] were used as ligands targeting the above-mentioned viral proteins. The plant metabolite structures were formatted as SYBYL mol2 file or in SMILES notation using Avogadro (version 1.20) and were added to the UCSF Chimera 1.14 platform [26].
Molecular docking simulations
Molecular docking experiments were carried out on UCSF Chimera 1.14 platform with AutoDock Vina plugged-in as docking algorithm [20]. Protein structures in three dimensions were opened in PDB formats. Co-crystallized ligands and other molecules were removed from the crystallized protein. Ligands were added in the platform as SYBYL mol2 files or in SMILES notation. Ligand and protein structures were minimized through addition of missing hydrogen atoms and charges to the structures using the Gasteiger charge method, which was computed using Amber’s Antechamber module [27]. ‘Flexible ligand into flexible active site’ protocol was followed during execution of docking procedures. In this protocol, flexible ligands were allowed and positioned within a grid box (Supplementary Table 2) which encompasses the enzymatic ligand-binding cavity, as predicted using COACH algorithms [28].
Druggability, ADME, and toxicity prediction
Top ten compounds per nsp, which in total were twenty-seven compounds, were selected for druggability, pharmacokinetic, and toxicity analyses. Absorption, distribution, metabolism, and excretion (ADME) properties of top twenty-seven compounds overall were computationally predicted using SwissADME software. Evaluation of pharmacokinetic profiles of compounds was performed according to Lipinski's ‘rule of five’ which assesses biochemical properties of a drug candidate involved in permeation and cell absorption. Three of the following values need to be met according to Lipinski’s criteria: < 500 Daltons (Da) for molecular weight, < 5 for calculated lipophilicity (MLogP), < 10 for the number of hydrogen-bond acceptors, and < 5 for the number of hydrogen bond donors [29]. Moreover, toxicity of hit compounds, specifically mutagenicity, tumorigenicity, reproductive toxicity, and irritant effects, were predicted in silico using OSIRIS Property Explorer software [30]. Solubility (LogS) was also predicted using the same software in which LogS ≥ − 4 indicates good solubility and favorable absorption of compounds.
Molecular dynamics simulations
Molecular dynamics (MD) simulation was employed to understand the dynamic behavior of the top-binding complexes based on molecular docking analysis. All MD simulations were carried out using GROMACS version 2020.1 under the Ubuntu Linux platform version 2020.1-1 [31]. The SARS-CoV-2 non-structural protein topologies were generated using the CHARMM 36 force field with TIP3P water model, while ligand topology was generated using CGenFF (CHARMM general force field). The complex was solvated on a dodecahedron grid by single point charge (SPC) water. The system was then neutralized with counterions. Energy minimization was done on the system using the steepest descent integrator for 5000 steps and Particle Mesh Ewald (PME) algorithm for the Coulomb and van der Waals interactions [32]. After system equilibration, each system was subjected to molecular dynamic simulation for 20 ns at constant temperature of 300 K. The dynamic trajectories were recorded during the production every 0.01 ns which were used to analyze the root mean square deviation (RMSD) and root mean square fluctuation (RMSF) for each system.
Results
One hundred four repurposed anti-HIV reverse transcriptase phytochemicals against SARS-CoV-2 nsps comprised of polyphenolics, terpenoids, alcohols, and alkaloids were docked with nsps3, 5, 10, 12, 13, 15, and 16. Twenty-seven compounds, which are included in the top 10 compounds per nsp, showed favorable binding affinities (Fig. 1). In addition, twenty of the top compounds exhibited multi-targeting properties.
Molecular docking with autolytic-processing enzymes (nsp3 and nsp5)
Top ten compounds against PLpro exhibited binding affinities of − 10.1 to − 10.8 kcal/mol (Table 1). The biflavonoid amentoflavone (1) exhibited highest affinity to PLpro with its benzopyrone (ring C) and phenolic moiety (ring B) participating through H-bonding (5.62 Å) and pi-anion interactions with Lys711, respectively (Fig. 2A). Ring C additionally bound Ile580 through pi-alkyl interaction. The phenolic functionality in ring B also participated in hydrogen bonding with His342 (3.49 Å) and in pi-alkyl binding with Ala579 and Leu742. Ring A’ of the benzopyrone moiety bound Arg712 by H-bonding (5.72 Å) and Ile310 by pi-alkyl interaction. Meanwhile, the phenolic moiety (ring B’) exhibited pi-anion interaction with Asp339 and pi-cation interaction with Arg558.

Top 1-binding compounds in complex with their target cysteine proteases: A amentoflavone (1) in complex with PLpro, B amentoflavone (1) (left) and volkensiflavone (11) (right) in complex with 3CLpro
On the other hand, top-ranked ligands against 3CLpro exhibited binding affinities of − 7.9 to − 8.6 kcal/mol (Table 1). The biflavones amentoflavone (1) and volkensiflavone (11) showed highest affinity to 3CLpro (Fig. 2B). The chromanone moieties (rings A′ and C) of amentoflavone showed stacked amide-pi and pi-pi T-shaped interactions with His41, a component of the 3CLpro catalytic dyad. These interactions were also demonstrated by its phenolic moiety to Asn142. The chromanone moiety (rings A and C) was bound to Met165 through pi-alkyl interaction along with hydrogen bonds with Val186 (5.46 Å), Arg188 (5.86 Å), and Glu166 (4.83 Å). Ring A′ also bound Cys44 through H-bonding (4.77 Å). Volkensiflavone (11) was likewise bound to the 3CLpro catalytic dyad, His41 and Cys145, through pi-anion interaction and hydrogen bonding (3.71 Å) of the chromanone moieties (rings C and A′ respectively). Moreover, rings A′ of the flavone substructure also exhibited hydrogen bonding with Glu166 (3.88 Å) while the B ring residue bound Thr25 through a pi-sigma interaction.
Molecular docking with replication-transcription complex enzymes (nsp12 and nsp13)
Among the top ten compounds against RdRp with binding affinities of − 8.6 to − 9.5 kcal/mol, the ellagitannin punicalin (9) exhibited the highest affinity (Table 2; Fig. 3A). The ellagic acid moiety occupied Ile494 and its galloyl hydroxyl bound Asn497 (4.13 Å), which are both components of the RdRp finger domain that is responsible for the entry and exit of the RNA template during replication-transcription [33]. Moreover, its glucose hydroxyl and hydrogen participated in hydrogen bonding (4.79 Å) and carbon-hydrogen bonding respectively with Asp684, a component of the motif B of the polymerase active site [34]. Other interactions include the participation of its ellagic acid moiety in pi-alkyl interaction with Lys577, galloyl hydroxyl in hydrogen bonding with Gly590 (2.87 Å), carbonyl oxygen in hydrogen bonding with Tyr689 (5.76 Å), and glucose moiety in carbon-hydrogen bonding with Ala685.

Top 1-binding compounds in complex with their target enzymes vital in replication: A punicalin (9) in complex with RdRp, B rhusflavanone (13) (left) and morelloflavone (14) (right) bound to helicase
Meanwhile, the top-ranked ligands against helicase had binding affinities of − 8.4 to − 9.2 kcal/mol in which the biflavonoids rhusflavanone (13) and morelloflavone (14) exhibited the strongest affinity (Fig. 3B). Compound 13 occupied the helicase Rec1A domain, which is a component of the nucleotide binding site, through hydrogen bonding of its chromanone (ring A′) hydroxyl and pyrone (ring C′) oxygen with Lys288 (4.76 Å) and Ala316 (3.32 Å), respectively [35]; pi-alkyl interactions of its chromene moieties, rings A with Ala316 with Ala316, and ring A′ with Lys320; and pi-cation interaction of its hydroxyphenyl moiety (ring B) with Lys320.
Compound 13 also occupied the Rec2A domain of the nucleotide binding site through a hydrogen bond of its pyrone (ring C) carbonyl with Arg443 (4.73 Å), an amide-pi stacked interaction of its hydroxyphenyl moiety (ring B′) with Gly538, and a pi-sigma interaction of ring B′ with Ser539. A pyrone (ring C) carbonyl further contributed to the binding affinity of compound 13 by binding to Thr286 through van der Waals forces. On the other hand, the dihydroxyphenyl moiety (ring B′) of compound 14 bound Glu341 through a relatively strong hydrogen bonding (3.02 Å) and both Ala312 and Val340 by pi-alkyl interactions. Ring A of its chromanone functionality bound Ala313 through a pi-alkyl interaction and also Ala312 by pi-sigma interaction. These residues are members of the helicase Rec2A domain of the nucleotide binding site. In addition, a benzophyrone hydroxyl (ring A′) of compound 14 bound Asp534 (5.32 Å), which is a residue of the Rec1A helicase domain of the nucleotide binding site.
Molecular docking with enzymes functioning in the evasion of host immunity
SAM-dependent 2′-O-methyltransferase complex enzymes (nsp16-nsp10 complex)
Top compounds against nsp16 exhibited affinities from − 9.3 to − 10.6 kcal/mol. The SAM-binding site was targeted and the biflavonoid robustaflavone (4) and the alkaloid michellamine B (19) demonstrated the greatest affinity (Table 3; Fig. 4A). Hydroxyphenyl ring B of compound 4 exhibited pi-alkyl interactions with Leu6898 and Met6929, and a hydrogen bonding with Cys6913 (3.64 Å). The hydroxyphenyl ring B′ showed carbon-hydrogen bonding with Asn6841. Moreover, its benzopyrone moiety (rings A and C) was in pi-anion interaction with Asp6897 and its chromene hydrogen was in hydrogen bonding with Asp6928 (3.40 Å). A van der Waals force interaction between its pyrone ring C oxygen and Gly6869 was also observed. Another pyrone moiety (ring C′) also interacted with nsp16 through a pi-anion interaction with Glu7001. Lys6844 (5.74 Å) and Asn6996 (4.96 Å) were occupied by the pyrone ring C′ carbonyl through hydrogen bonding. On the other hand, compound 19’s isoquinoline moiety was in H-bonding with Asp6928 (3.68 Å) and Asp6897 (4.60 Å) and in carbon-hydrogen bonding with Gly6869. Another isoquinoline moiety was in pi-anion interaction with Asp6931. Moreover, the naphthalene moiety participated in pi-pi T-shaped interaction with Phe6947 and in pi-sulfur interaction with Cys6914. The methyl group connected to naphthalene manifested alkyl interactions with Met6929, Leu6898, and Cys6913.

Top 1-binding compounds in complex with nsps involved in host immunity evasion: A robustaflavone (4) (left) and michellamine B (19) (right) complexed with S-adenosylmethionine-dependent 2′-O-methyltransferase, B robustaflavone (4) bound to nsp10, and C hinokiflavone (15) bound to nsp15
In connection, top compounds against nsp10 showed binding affinities of − 6.9 to − 7.7 kcal/mol. The interface between nsp10 and nsp16 was targeted and several interactions were observed. Biflavonoid robustaflavone (4) had the highest affinity (Fig. 4B). Its pyrone ring C′ was in carbon-hydrogen bonding with Ile4334. Chromanone (ring A′) hydroxyl formed a strong H-bonding with Asp4335 (3.50 Å). Carbon atoms of chromanone (rings A and C) and hydroxyphenyl ring B′ formed salt bridges with Lys4346 while ring C′ carbonyl exhibited a salt bridge with Arg4331.
Endoribonuclease (nsp15)
Top-scoring compounds against nsp15 exhibited affinities of − 8.6 to − 7.3 kcal/mol, stronger (Table 4). The biflavonone hinokiflavone (15) scored the highest affinity, noting its interactions with its putative binding site that is proximal to the catalytic triad of His235, His250, and Lys290: flavone moiety (rings A, B, and C) in hydrogen bonding (4.55 Å) and pi-alkyl interaction with Met243, pyrone ring C in pi-sigma interaction with Tyr262, pyrone ring C′ in pi-anion interaction with Glu258, and hydroxyphenyl ring B′ in pi-alkyl and pi-pi stacked interactions with Ala256 and His362, respectively (Fig. 4C).
Druggability, ADME, and toxicity
Six of the 20 top-scoring and multi-targeting repurposed phytochemicals were found to be druggable according to Lipinski’s rule of five (Table 5). Hinokiflavone (15) is a top-scoring, multi-targeting, druggable compound. Moreover, compounds 5 and 17 were multi-targeting and exhibited good gastrointestinal absorption properties.
In addition, compounds 25 and 26 showed the best solubility in water of − 2.85, thereby depicting good excretion properties (Table 6). Toxicity prediction through OSIRIS Property Explorer showed that all top compounds except 11, 14, 15, 18, 19, and 20 have no mutagenic, tumorigenic, irritant, and reproductive toxicity risks (Table 6).
Molecular dynamics simulations
Molecular dynamics (MD) simulation was performed on the top-binding ligands, chosen based on molecular docking and ADMET analyses, to assess at an atomic level the binding behavior of the various polyphenols against SARS-CoV-2 non-structural proteins. The stability of the complexes, specifically PLpro-amentoflavone (1), 3CLpro-amentoflavone (1), RdRp-punicalin (9), helicase-rhusflavanone (13), nsp16-michellamine B (19), nsp10-robustaflavone (4), and nsp15-hinokiflavone (15), was evaluated using post-simulation parameters root mean square deviation (RMSD) and root mean square fluctuations (RMSF). RMSD is one of the widely used analyses using MD trajectories of protein-ligand complexes to establish equilibrium within a given simulation period. Based on RMSD analysis which was measured as an average throughout a 20-ns simulation, the complexes attained dynamic stability (Fig. 5). In the case of the amentoflavone (1)-bound PLpro, it took some time for the complex to reach equilibrium. As shown in the plot of RMSD (Å) versus simulation time (ns), a steady increase in RMSD can be observed up to 15 ns before stabilization occurs. A similar trend can be observed for the RdRp-punicalin (9) complex. After a steady rise in RMSD, the complex achieved equilibrium around 13 ns with an average RMSD value of 7.1 Å. Among the complexes, the amentoflavone (1)-bound 3CLpro appeared to be the most stable complex having the lowest average equilibrium RMSD (2.2 Å). Although a minor fluctuation can be observed around 11 ns, the complex remained stable for the entire simulation time. In the case of nsp16-michellamine B (19) complex, an incremental increase in RMSD can be noted from the start of the simulation until 8 ns and a relatively high divergence can be seen around 9 ns. However, the system gained equilibrium thereafter. For the helicase-rhusflavanone (13) complex, several minor fluctuations can be noticed from initial binding stage up to mid simulation time. Despite this observation, the average RMSD of the complex remained low (2.8 Å). Another relatively stable complex is robustaflavone (4) bound to nsp10 with an average equilibrium RMSD of 3.9 Å. Shortly after ligand-binding, the complex attained equilibrium. Lastly, the nsp15-hinokiflavone (15) complex exhibited relatively low stability based on the RMSD plot where some minor fluctuations are noted at the beginning of the simulation up to 15 ns.

Root mean square deviation (RMSD, Å) as a function of time of the top-binding complexes
The time-averaged residual fluctuations of the seven top-binding complexes were also analyzed on the basis of trajectory data within a 20-ns simulation (Fig. 6). The results of the residual RMSF analysis revealed that most of the complexes, particularly 3CLpro-amentoflavone (1), nsp16-michellamine B (19), nsp10-robustaflavone (4), helicase-rhusflavanone (13), have average RMSF values ranging from 1.1 to 1.6 Å and have shown relatively stable fluctuation patterns. These data are consistent with RMSD analysis, which confirm that the said complexes are stable. For the hinokiflavone (15)-bound nsp15 complex, the average residual fluctuations are slightly higher (2.2 Å). For the larger protein complexes—RdRP and PLpro, higher fluctuations were observed, averaging 3.3 Å and 3.5 Å, respectively. Such an RMSF pattern for the two complexes justifies the observed longer time for stabilization to occur during RSMD calculations.

Time-averaged root mean square fluctuations (RMSF, Å) as a function amino acid residue sequence number of the top-binding complexes
Discussion
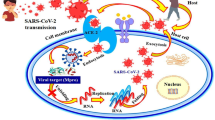
The SARS-CoV-2 non-structural proteins (nsps) play vital roles in the virus’ pathogenesis, survival, and virulence. A number of these nsps have been considered as attractive and important drug targets due to their involvement in viral post-translational processing, replication, and host immunity evasion mechanisms (Fig. 7). The cysteine proteases, nsp3 (PLpro) and nsp5 (3CLpro), are involved in the autolytic cleavage of the polyproteins pp1a and pp1ab wherein PLpro cleaves 3 sites at the N-terminus while 3CLpro cleaves through the remaining sites (11 sites in pp1ab) to release the nsps [36]. Proceeding to the replication-transcription complex, nsp12 (RdRp) elongates the daughter strand through the polymerization of nucleotides while nsp13 (helicase) clears RNA secondary structures and RNA-binding proteins [37]. The nsp 16 (SAM-dependent 2′-O-methyltransferase) in complex with nsp10 as its cofactor provides a 5′ cap to the RNA genome through C2′-O-methyl-ribosyladenine, conferring RNA stability and host cell immunity protection [38]. Lastly, the nsp15 (endoribonuclease) hinders recognition of dsRNA intermediates by host dsRNA sensors [33]. Our results, therefore, highlight the role of anti-HIV RT phytochemicals as potential antagonists of SARS-CoV-2 by interfering with the discussed mechanisms.

The SARS-CoV-2 life cycle highlighting the role of nsps in replication and transcription and the potential inhibited target SARS-CoV-2 nsps of repurposed anti-HIV RT phytochemicals. SARS-CoV-2 virion image credit: CDC/ Alissa Eckert (MSMI) and Dan Higgins (MAMS)
Natural products have been a subject of investigation concerning their ability to antagonize SARS-CoV-2 due to their availability and wide range of health benefits [34, 35, 39,40,41]. In relation, repurposing established anti-HIV phytochemicals means that the lead compounds in this study can be easily obtained from previously explored plants that are consumed by populations. Here, we focused on the employment of computational target-based drug discovery methodologies, such as molecular docking, molecular dynamic simulations, and pharmacokinetic property predictions in search for potential hits for inhibiting the aforementioned SARS-CoV-2 nsps. Our study revealed that the biflavonoid amentoflavone (1) showed the highest binding to both SARS-CoV-2 cysteine proteases PLpro and 3CLpro. Compound 1, isolated from the Chinese olive fruit, Canarium album [42], was also reported in a previous study to be a potent inhibitor of SARS-CoV PLpro [43, 44]. Volkensiflavone (11) from the seeds and rinds of Garcinia intermedia [45] was another top compound against 3CLpro. Punicalin (9) from the pomegranate Punica granatum peel [46] exhibited high binding propensity against RdRp, an enzyme considered to be a promising target inhibiting viral replication. Morelloflavone (14) from G. intermedia was first to be reported here with an inhibitory potential against SARS-CoV-2 helicase. Interestingly, its potential extends against 3CLpro [47]. Robustaflavone (4) from the leaves of Garcinia epunctata [48] showed the best potential against the 2′-O-methyltransferase and its cofactor. This is the first investigation of its activity against these nsps aside from its interaction with 3CLpro [49]. On the other hand, michellamine B (19) from Ancistrocladus korupensis leaves [50] manifested an inhibitory potential against nsp16, therefore opening the doors of phenolic alkaloids against SARS-CoV-2. Lastly, hinokiflavone (15) from Selaginella tamariscina [51] was reported to be a potential 3CLpro inhibitor and potent against the replication-transcription complex [43, 47, 49, 52]. This, however, is the first investigation of its activity against the endoribonuclease of SARS-CoV-2 in silico. The multi-targeting potential of some of these compounds increases the chance of getting a maximal inhibitory effect [53].
To further validate the molecular docking analysis, the top-binding ligands were submitted for molecular dynamic simulations. Through post-simulation analyses, the top-binding ligands were generally found to be dynamically stable upon binding to respective proteins. Although most of the top 1 compounds were predicted in silico to be non-druggable, efforts are rising to explore compounds in the oral druggable space beyond the rule of five (bRo5) [54, 55]. Additionally, four of these did not manifest toxicity in silico. The biflavonoids volkensiflavone (11), morelloflavone (14), and hinokiflavone (15) were computationally predicted as non-mutagenic, non-tumorigenic, and non-irritant, but were predicted to pose reproductive toxicity risk which may be attributed to their chromene and hydroxyphenyl moieties. It should be noted, however, that hinokiflavone (15) is a druggable top 1 compound. Michellamine B (19) was predicted to be tumorigenic due to its naphthalene moiety. In addition, compounds 5 and 17 exhibited good gastrointestinal absorptive features as implicated by their favorable lipophilicity and polar surface area [56]. These also did not manifest any form of toxicity in silico. Despite computational incompatibilities, these compounds can still serve as templates for drug design and undergo in vitro and in vivo assays for validating their anti-SARS-CoV-2 properties, noting that their promising polyphenolic nature allowed them to form hydrogen bonds with key residues of the SARS-CoV-2 nsps. With the validation of pre-clinical experiments, the secondary metabolites can be produced through in vitro plant tissue cultures that can be augmented by metabolic engineering, elicitation, and even the use of bioreactors [57, 58].
Conclusions
The search for anti-COVID-19 therapeutic agents is a response to the continuous spread of the virus amidst vaccine availability. The similarity between the pathogenesis of HIV and SARS-CoV-2 inspired the repurposing of previously reported anti-HIV reverse transcriptase phytochemicals against SARS-CoV-2 nsps implicated in viral replication, post-translational processing, and host immunity evasion mechanisms. The top-ranking polyphenolics amentoflavone (1), robustaflavone (4), punicalin (9), volkensiflavone (11), rhusflavanone (13), morelloflavone (14), hinokiflavone (15), and michellamine B (19) can be further screened using confirmatory in vitro and in vivo assays, and can serve as prototypes for designing novel anti-COVID-19 drugs in consideration of their polyphenolic nature. As promising drug templates, functionalities in the compound structure can be modified to improve druggability and pharmacokinetic properties.
Availability of data and materials
All data generated in this study are included in this published article and the supplementary information files.
Abbreviations
- 3CLpro:
-
3-Chymotrypsin-like protease
- ADME:
-
Absorption, digestion, metabolism, and excretion
- BE:
-
Binding energy
- COVID-19:
-
Coronavirus disease 2019
- Cpd:
-
Compound
- dsDNA:
-
Double-stranded DNA
- HIV:
-
Human immunodeficiency virus
- kcal/mol:
-
Kilocalorie per mole
- nsp:
-
Non-structural protein
- PDB:
-
Protein data bank
- PLpro:
-
Papain-like protease
- RdRp:
-
RNA-dependent RNA polymerase
- RNA:
-
Ribonucleic acid
- RT:
-
Reverse transcriptase
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus 2
References
Guo Q, Xu W, Wang P-F, Ji H-Y, Zhang X-L, Wang K, Li J (2021) Facing coronavirus disease 2019: What do we know so far? (Review). Exp Ther Med 21(6):658–663. https://doi.org/10.3892/etm.2021.10090
Wang C, Xiao X, Feng H, Hong Z, Li M, Tu N, Li X, Wang K, Bu L (2021) Ongoing COVID-19 pandemic: a concise but updated comprehensive review. Curr Microbiol 78(5):1718–1729. https://doi.org/10.1007/s00284-021-02413-z
World Health Organization. WHO coronavirus disease (COVID-19) dashboard. https://covid19.who.int/. Accessed 26 May 2021.
Astuti I, Ysrafil (2020) Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2): An overview of viral structure and host response. Diabetes Metab Syndr 14:407-412. https://doi.org/10.1016/j.dsx.2020.04.020.
Shahzad F, Anderson D, Najafzadeh M (2020) The antiviral, anti-inflammatory effects of natural medicinal herbs and mushrooms and SARS-CoV-2 infection. Nutrients 12(9). https://doi.org/10.3390/nu12092573
Pandey A, Khan MK, Hamurcu M, Gezgin S (2020) Natural plant products: a less focused aspect for the COVID-19 viral outbreak. Front Plant Sci 11. 11. https://doi.org/10.3389/fpls.2020.568890
Forrestall K, Burley D, Cash M, Pottie I, Darvesh S (2020) 2-Pyridone natural products as inhibitors of SARS-CoV-2 main protease. Chem Biol Interact 335:109348. https://doi.org/10.1016/j.cbi.2020.109348
Notarte KI, Devanadera MK, Mayor AB, Cada MC, Pecundo MH, Macabeo AP (2019) Toxicity, antibacterial, and antioxidant activities of fungal endophytes Colletotrichum and Nigrospora spp. isolated from Uvaria grandiflora. Philipp J Sci 148:503–510
Quimque MT, Notarte KI, Letada A, Fernandez RA, Pilapil DY, Pueblos KR et al. (2021) Potential cancer- and Alzheimer's disease-targeting phosphodiesterase inhibitors from Uvaria alba: Insights from in vitro and consensus virtual screening. ACS Omega 6(12):8403–8417. https://doi.org/10.1021/acsomega.1c00137
Paraiso IL, Revel JS, Stevens JF (2020) Potential use of polyphenols in the battle against COVID-19. Curr Opin Food Sci 32:149–155. https://doi.org/10.1016/j.cofs.2020.08.004
Levy E, Delvin E, Marcil V, Spahis S (2020) Can phytotherapy with polyphenols serve as a powerful approach for the prevention and therapy tool of novel coronavirus disease 2019 (COVID-19)? Am J Physiol Endocrinol Metab 319(4):E689–E708. https://doi.org/10.1152/ajpendo.00298.2020
Annunziata G, Sanduzzi Zamparelli M, Santoro C, Ciampaglia R, Stornaiuolo M, Tenore GC, Sanduzzi A, Novellino E (2020) May polyphenols have a role against coronavirus infection? An overview of in vitro evidence. Front Med (Lausanne) 7. https://doi.org/10.3389/fmed.2020.00240
Mehany T, Khalifa I, Barakat H, Althwab SA, Alharbi YM, El-Sohaimy S (2021) Polyphenols as promising biologically active substances for preventing SARS-CoV-2: a review with research evidence and underlying mechanisms. Food Biosci 40:100891. https://doi.org/10.1016/j.fbio.2021.100891
Benarba B, Pandiella A (2020) Medicinal plants as sources of active molecules against COVID-19. Front Pharmacol 11. https://doi.org/10.3389/fphar.2020.01189
Boukhatem MN, Setzer WN (2020) Aromatic herbs, medicinal plant-derived essential oils, and phytochemical extracts as potential therapies for coronaviruses: Future perspectives. Plants (Basel) 9. 9(6). https://doi.org/10.3390/plants9060800
Quimque MTJ, Notarte KIR, Fernandez RAT, Mendoza MAO, Liman RAD, Lim JAK, Pilapil LAE, Ong JKH, Pastrana AM, Khan A, Wei DQ, Macabeo APG (2020) Virtual screening-driven drug discovery of SARS-CoV2 enzyme inhibitors targeting viral attachment, replication, post-translational modification and host immunity evasion infection mechanisms. J Biomol Struct Dyn 16:1–18. https://doi.org/10.1080/07391102.2020.1776639
Gogoi M, Borkotoky M, Borchetia S, Chowdhury P, Mahanta S, Barooah AK (2021) Black tea bioactives as inhibitors of multiple targets of SARS-CoV-2 (3CLpro, PLpro and RdRp): a virtual screening and molecular dynamic simulation study. J Biomol Struct Dyn 10:1–24. https://doi.org/10.1080/07391102.2021.1897679
Stasiulewicz A, Maksymiuk AW, Nguyen ML, Bełza B, Sulkowska JI (2021) SARS-CoV-2 papain-like protease potential inhibitors-In silico quantitative assessment. Int J Mol Sci 22(8):3957–3986. https://doi.org/10.3390/ijms22083957
Ghazwani MY, Bakheit AH, Hakami AR, Alkahtani HM, Almehizia AA (2021) Virtual screening and molecular docking studies for discovery of potential RNA-dependent RNA polymerase inhibitors. Crystals 11(5):471. https://doi.org/10.3390/cryst11050471
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera-A visualization system for exploratory research and analysis. J Comput Chem 25(13):1605–1612. https://doi.org/10.1002/jcc.20084
Yoshimoto FK (2020) The proteins of severe acute respiratory syndrome coronavirus-2 (SARS CoV-2 or n-COV19), the cause of COVID-19. Protein J 39(3):198–216. https://doi.org/10.1007/s10930-020-09901-4
Gorbalenya AE, Koonin EV, Donchenko AP, Blinov VM (1989) Coronavirus genome: prediction of putative functional domains in the non-structural polyprotein by comparative amino acid sequence analysis. Nucleic Acids Res 17(12):4847–4861. https://doi.org/10.1093/nar/17.12.4847
Seybert A, Hegyi A, Siddell SG, Ziebuhr J (2000) The human coronavirus 229E superfamily 1 helicase has RNA and DNA duplex-unwinding activities with 5'-to-3' polarity. RNA 6(7):1056–1068. https://doi.org/10.1017/s1355838200000728
van Dinten LC, van Tol H, Gorbalenya AE, Snijder EJ (2000) The predicted metal-binding region of the arterivirus helicase protein is involved in subgenomic mRNA synthesis, genome replication, and virion biogenesis. J Virol 74(11):5213–5223. https://doi.org/10.1128/jvi.74.11.5213-5223.2000
Chinsembu KC (2019) Chemical diversity and activity profiles of HIV-1 reverse transcriptase inhibitors from plants. Rev Bras Farmacogn 29(4):504–528. https://doi.org/10.1016/j.bjp.2018.10.006
Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E, Hutchison GR (2012) Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J Cheminformatics 4. 4(1). https://doi.org/10.1186/1758-2946-4-17
Wang J, Wang W, Kollman PA, Case DA (2006) Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Graph Model 25(2):247–260. https://doi.org/10.1016/j.jmgm.2005.12.005
Yang J, Roy A, Zhang Y (2013) Protein-ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 29(20):2588–2595. https://doi.org/10.1093/bioinformatics/btt447
Macabeo APG, Cruz AJC, Narmani A, Arzanlou M, Babai-Ahari A, Pilapil LAE, Garcia KYM, Huch V, Stadler M (2020) Tetrasubstituted α-pyrone derivatives from the endophytic fungus, Neurospora udagawae. Phytochem Lett 35:147–151. https://doi.org/10.1016/j.phytol.2019.11.010
Phukhamsakda C, Macabeo APG, Huch V, Cheng T, Hyde KD, Stadler M (2019) Sparticolins A-G, biologically active oxidized spirodioxynaphthalene derivatives from the ascomycete Sparticola junci. J Nat Prod 82(10):2878–2885. https://doi.org/10.1021/acs.jnatprod.9b00604
Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ (2005) GROMACS: Fast, flexible, and free. J Comput Chem 26(16):1701–1718. https://doi.org/10.1002/(ISSN)1096-987X
Toukmaji A, Sagui C, Board J, Darden T (2000) Efficient particlemesh ewald based approach to fixed and induced dipolar interactions. J Chem Phys 113(24):10913–10927. https://doi.org/10.1063/1.1324708
Deng X, Hackbart M, Mettelman RC, O'Brien A, Mielech AM, Yi G, Kao CC, Baker SC (2017) Coronavirus nonstructural protein 15 mediates evasion of dsRNA sensors and limits apoptosis in macrophages. Proc Natl Acad Sci USA 114(21):E4251–E4260. https://doi.org/10.1073/pnas.1618310114
Kim Y, Wower J, Maltseva N, Chang C, Jedrzejczak R, Wilamowski M, Kang S, Nicolaescu V, Randall G, Michalska K, Joachimiak A (2021) Tipiracil binds to uridine site and inhibits Nsp15 endoribonuclease NendoU from SARS-CoV-2. Commun Biol 4(1):193. https://doi.org/10.1038/s42003-021-01735-9
Antonio AS, Wiedemann LSM, Veiga-Junior VF (2020) Natural products’ role against COVID-19. RSC Adv 10(39):23379–23393. https://doi.org/10.1039/D0RA03774E
Wu C, Liu Y, Yang Y, Zhang P, Zhong W, Wang Y, Wang Q, Xu Y, Li M, Li X, Zheng M, Chen L, Li H (2020) Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm Sin B 10(5):766–788. https://doi.org/10.1016/j.apsb.2020.02.008
Chen J, Malone B, Llewellyn E, Grasso M, Shelton PMM, Olinares PDB, Maruthi K, Eng ET, Vatandaslar H, Chait BT, Kapoor TM, Darst SA, Campbell EA (2020) Structural basis for helicase-polymerase coupling in the SARS-CoV-2 replication-transcription complex. Cell 182(6):1560–1573. https://doi.org/10.1016/j.cell.2020.07.033
Rosas-Lemus M, Minasov G, Shuvalova L, Inniss NL, Kiryukhina O, Brunzelle J, Satchell KJF (2020) High-resolution structures of the SARS-CoV-2 2′-O-methyltransferase reveal strategies for structure-based inhibitor design. Sci Signal 13. 13(651):eabe1202. https://doi.org/10.1126/scisignal.abe1202
Boozari M, Hosseinzadeh H (2021) Natural products for COVID-19 prevention and treatment regarding to previous coronavirus infections and novel studies. Phytother Res 35(2):864–876. https://doi.org/10.1002/ptr.6873
Huang J, Tao G, Liu J, Cai J, Huang Z, Chen JX (2020) Current prevention of COVID-19: natural products and herbal medicine. Front Pharmacol 11. https://doi.org/10.3389/fphar.2020.588508
Menezes JCJMDS, Campos VR (2021) Natural biflavonoids as potential therapeutic agents against microbial diseases. Sci Total Environ 769:145168. https://doi.org/10.1016/j.scitotenv.2021.145168
He Z, Xia W, Chen J (2008) Isolation and structure elucidation of phenolic compounds in Chinese olive (Canarium album L.) fruit. Eur Food Res Technol 226(5):1191–1196. https://doi.org/10.1007/s00217-007-0653-5
Eydoux C, Fattorini V, Shannon A, Le TT, Didier B, Canard B, Guillemot JC (2021) A fluorescence-based high throughput-screening assay for the SARS-CoV RNA synthesis complex. A fluorescence-based high throughput-screening assay for the SARS-CoV RNA synthesis complex. J Virol Methods 288:288. https://doi.org/10.1016/j.jviromet.2020.114013
Ryu YB, Jeong HJ, Kim JH, Kim YM, Park JY, Kim D, Nguyen TT, Park SJ, Chang JS, Park KH, Rho MC, Lee WS (2010) Biflavonoids from Torreya nucifera displaying SARS-CoV 3CL(pro) inhibition. Bioorg Med Chem 18(22):7940–7947. https://doi.org/10.1016/j.bmc.2010.09.035
Acuña UM, Figueroa M, Kavalier A, Jancovski N, Basile MJ, Kennelly EJ (2010) Benzophenones and biflavonoids from Rheedia edulis. J Nat Prod 73(11):1775–1779. https://doi.org/10.1021/np100322d
Singh B, Singh JP, Kaur A, Singh N (2018) Phenolic compounds as beneficial phytochemicals in pomegranate (Punica granatum L.) peel: a review. Food Chem 261:75–86. https://doi.org/10.1016/j.foodchem.2018.04.039
Teli DM, Shah MB, Chhabria MT (2021) In silico screening of natural compounds as potential inhibitors of SARS-CoV-2 main protease and spike RBD: Targets for COVID-19. Front Mol Biosci 7. https://doi.org/10.3389/fmolb.2020.599079
Emade Kwene C, Tih AE, Abderamane B, Ghogomu RT (2020) Two new phenolic glycosides from the leaves of Garcinia epunctata Stapf. Z Naturforsch C J Biosci 75(1-2):51–56. https://doi.org/10.1515/znc-2018-0217
Lokhande K, Nawani N, K Venkateswara S, Pawar S (2020) Biflavonoids from Rhus succedanea as probable natural inhibitors against SARS-CoV-2: a molecular docking and molecular dynamics approach [pre-print]. J Biomol Struct Dyn 1–13. https://doi.org/10.1080/07391102.2020.1858165
McMahon JB, Currens MJ, Gulakowski RJ, Buckheit RW, Jr Lackman-Smith C, Hallock YF et al (1995) Michellamine B, a novel plant alkaloid, inhibits human immunodeficiency virus-induced cell killing by at least two distinct mechanisms. Antimicrob Agents Chemother 39(2):484–488. https://doi.org/10.1128/aac.39.2.484
Zhang GG, Jing Y, Zhang HM, Ma EL, Guan J, Xue FN, Liu HX, Sun XY (2012) Isolation and cytotoxic activity of selaginellin derivatives and biflavonoids from Selaginella tamariscina. Planta Med 78(04):390–392. https://doi.org/10.1055/s-0031-1298175
Ngo ST, Pham NQA, Le LT, Pham D-H, Vu VV (2020) Computational determination of potential inhibitors of SARS-CoV-2 main protease. J Chem Inf Model 60(12):5771–5780. https://doi.org/10.1021/acs.jcim.0c00491
Senanayake SL (2020) Overcoming nonstructural protein 15-nidoviral uridylate-specific endoribonuclease (nsp15/NendoU) activity of SARS-CoV-2. Future Drug Discov 2(3). https://doi.org/10.4155/fdd-2020-0012
Doak BC, Over B, Giordanetto F, Kihlberg J (2014) Oral druggable space beyond the rule of 5: insights from drugs and clinical candidates. Chem Biol 21(9):1115–1142. https://doi.org/10.1016/j.chembiol.2014.08.013
Tyagi M, Begnini F, Poongavanam V, Doak BC, Kihlberg J (2020) Drug syntheses beyond the rule of 5. Chemistry 26(1):49–88. https://doi.org/10.1002/chem.201902716
Daina A, Zoete V (2016) A BOILED-Egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem 11(11):1117–1121. https://doi.org/10.1002/cmdc.201600182
Bhojwani SS, Dantu PK (eds) (2013) Plant tissue culture: an introductory text. Springer, India. https://doi.org/10.1007/978-81-322-1026-9
Pant B (2014) Application of plant cell and tissue culture for the production of phytochemicals in medicinal plants. In: Adhikari R, Thapa S (eds) Infectious diseases and nanomedicine II, Advances in experimental medicine and biology, vol 808. Springer, New Delhi, pp 25–39. https://doi.org/10.1007/978-81-322-1774-9_3
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
VNODL, JAHM, DYHP: conceptualization, investigation, data collection and analysis, manuscript preparation, and manuscript editing. RATF: conceptualization, investigation, data collection and analysis, manuscript preparation, and manuscript editing and review. JKARC: data collection. MTJQ: conceptualization, investigation, data collection and analysis, manuscript editing, and manuscript review. JCMA: investigation and manuscript editing. KIRN: conceptualization, investigation, manuscript editing and manuscript review. APGM: conceptualization, design, manuscript preparation, editing, and review. All authors have read and approved the present version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
de Leon, V.N.O., Manzano, J.A.H., Pilapil, D.Y.H. et al. Anti-HIV reverse transcriptase plant polyphenolic natural products with in silico inhibitory properties on seven non-structural proteins vital in SARS-CoV-2 pathogenesis. J Genet Eng Biotechnol 19, 104 (2021). https://doi.org/10.1186/s43141-021-00206-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43141-021-00206-2