
Abstract
Background
Plant extracts have attracted significant interest among researchers due to their potential bioactivity and crucial contribution to the production of pharmaceutical compounds. In this study, the primary objective was to extract, analyze and characterize the bioactive compounds found in the methanol root extract of Datura fastuosa (D. fastuosa). This was achieved using various analytical techniques such as gas chromatography/mass spectrometry (GC–MS), ultra-violet visible spectrophotometry, Fourier-transform infrared (FT-IR), nuclear magnetic radiation spectrometry (NMR) and DPPH free radical scavenging activity assay.
Results
GC–MS analysis of the methanol root crude extract identified 49 compounds. Three compounds were isolated via column chromatography; one was pure, with a sharp melting point and clean IR spectrum, while the other two showed broad melting points and IR interferences. Comprehensive investigation of the pure extract revealed a UV profile with two distinct bands (300–800 nm) and confirmed functional groups (alcohol, alkanes, alkenes, carbonyl, methylene, and methyl) through FT-IR analysis. The 1HNMR (proton nuclear magnetic resonance spectroscopy) signal confirmed the presence of forty-nine non-equivalent protons, 13CNMR (Carbon-13 nuclear magnetic resonance spectroscopy) signal confirmed the presence of 32 non-equivalent carbons and DEPT-135 (distortionless enhancement by polarization transfer-135) signal confirmed the presence of 24 carbons (17 for odd and 7 for even) which are protons containing carbons in the compound. Combining the above mentioned analyses with data obtained from the GC/MS analysis of National Institute of Standards and Technology (NIST) library, the isolated pure compound exhibited a structural similarity to 1-(7-(3-hydroxyphenyl)-1,1,4a,5,6,9,10a,10b-octamethyl-1,2,3,4,4a,4b,6a,7,8,9,10,10a,10b,11,12,12a-hexadecahydrochrysen-2-yl)propan-1-one, with a chemical formula of C35H50O2.
Conclusions
The presence of various notable compounds, including phenolics, flavonoids, alkaloids, steroids etc., within the methanol root extract of D. fastuosa signifies its pharmacological potential. The methanol crude extract demonstrated antioxidant potential compared to standard ascorbic acid, exhibiting DPPH scavenging activity. Previous research has demonstrated the bioactivity of some of these compounds, further elucidating the plant’s medicinal properties. These findings not only suggest opportunities for developing synthetic drugs but also underscore its direct therapeutic potential in addressing diverse ailments.
Similar content being viewed by others

Background
Numerous plant species are known to contain bioactive compounds, referred to as secondary metabolites, which are synthesized within the plants and are primarily responsible for the diverse and distinct activities observed in plant extracts [1,2,3,4,5]. These bioactive compounds, including flavonoids, tannins, phenols, alkaloids, etc. have demonstrated the potential to treat various illnesses [6,7,8,9]. Throughout history, many cultures have relied on these medicinal plants to treat various illnesses, and even in the present time, with modern healthcare services available, numerous individuals, especially in developing countries, continue to depend on medicinal plants to address their primary healthcare needs. Moreover, natural products derived from plants and their structural analogs have played a significant role in advancing pharmaceuticals. Some pharmaceutical companies rely on these plant products for drug synthesis, with approximately 50% of drugs produced worldwide being based on these plant extracts [10, 11]. The medicinal plant investigated in this study is known as Datura fastuosa, which has several common names, including Devil’s trumpet and thorn apple. It belongs to the family Solanaceae and considered to be a poisonous plant when consumed in large doses [12,13,14]. However, it is also one of the most important medicinal herbs globally, renowned for its anti-inflammatory properties [14, 15]. Traditionally, it has been used in various medicinal practices to alleviate pain, breathlessness, and fever, and it has demonstrated antibacterial and antifungal activities against pathogens. The therapeutic benefits are predominantly associated with the presence of a diverse range of bioactive compounds found within Datura species. Although the quantity and composition of these constituents vary among different species, they typically encompass phytochemicals such as alkaloids, flavonoids, phenols, tannins, saponins, and sterols [16, 17]. Further identification and characterization of bioactive compounds in various species of these plants will prove beneficial. Datura fastuosa is commonly found in Africa, particularly in Southern Ethiopia, as well as in other tropical and subtropical regions. In the region of Konso, Southern Ethiopia, D. fastuosa is locally known as “Qorramakka”, and it is traditionally applied for treating snake venom in humans or animals and used as a snake and insect repellent. The plant typically grows up to 92 cm, and its leaves are covered with short and soft grayish hairs, measuring 10–20 cm in length and 5–18 cm in breadth [18]. The current study focused on exploring the bioactive phytochemicals within the methanolic root extract of D. fastuosa through a variety of techniques. These methods include gas chromatography–mass spectrometry (GC/MS) to ascertain the presence of bioactive substances. Additionally, the isolation of pure compounds from the crude extract were achieved using chromatographic methods, followed by structural analysis of the extracts through a combination of techniques, including UV, FT-IR, 1H-NMR, 13C-NMR, and DEPT-135 spectroscopy. To interpret the results, the acquired data was compared with reference data from the GC/MS analysis in the NIST Library.
Result
Sample extraction from root of Datura fastousa using different solvents
The crude chemical compounds present in D. fastousa roots were isolated using methanol, acetone, ethyl acetate, and chloroform. The yields of these extracts obtained through maceration. The percentage of extractable compounds contained in roots by using methanol, acetone, ethyl acetate, and chloroform are 4.9%, 0.61%, 0.39%, and 0.54% (w/w), respectively. Notably, methanol extraction exhibited the highest yield of crude extracts among the solvents tested. Consequently, the methanol root extract was chosen for further investigation.
Preliminary phytochemical screening
In this investigation, standard phytochemical screening procedures were employed to analyze the concentrated yield obtained through methanol-based extraction. The aim was to identify secondary metabolites present in the extract. Our findings revealed the significant abundance of these phytochemicals within the roots extracts of the D. fastousa plant. Specifically, the root extract of D. fastousa was found to contain varying levels of alkaloids, flavonoids, phenols, tannins, saponins, and steroids. Alkaloids, flavonoids, phenols, and saponins were notably prominent, while tannins and steroids were present in relatively lower concentrations. Interestingly, terpenoids and triterpenoids were absent in the extract (See Table 1).
DPPH free radical scavenging activity of the crude extracts
All extracts demonstrated antioxidant potential compared to standard ascorbic acid, with DPPH scavenging activity values ranging significantly (P < 0.05) from 12.5 to 400 mg/ml concentration. The methanol extract exhibited the greatest antioxidant potential, outperforming acetone, ethyl acetate, and chloroform extracts. The antioxidant activity of the root extract varied with the solvent used, with methanol being the most effective, followed by acetone, ethyl acetate, and chloroform, the least effective (see Table 2.)
Gas chromatography–mass spectrometry (GC/MS) analysis
In the study, GC/MS analysis was utilized on the methanol root extracts, revealing the presence of numerous bioactive compounds previously documented. This analysis clearly identified a total of 49 potentially bioactive compounds within the methanol root extract of D. fastousa (see Fig. 1). The identification of the phytochemical compounds was confirmed based on the retention time, molecular formula, molecular weight and % peak area. To elucidate these findings, we integrated the peaks observed in the chromatogram and cross-referenced them with the spectra of well-known compounds stored in the GC–MS NIST library database.

GC/MS spectrum of Datura fastuosa methanolic root extract
In this analysis, various molecules and compounds were identified through their retention time, molecular formula, molecular weight, peak area percentage, IUPAC (International Union of Pure and Applied Chemistry) name, and structure. These findings are presented in Table 3. The relative abundance displayed in this table represents the area under the peak as a percentage of the total area or as a percentage of the total height of all chromatographic peaks. Among the identified molecules or compounds, the following exhibit relatively high peak areas in the extract: 1-methyl-pyrrolidine-2-carboxylic acid, 8-mzabicyclo[3.2.1]octan-3-ol,8-methyl-,endo-,4-bromo-7-chloro-8-fluoro-2-phenylquinoline, atropine, [2,3-diacetyloxy-5-(2,4,6-triacetyloxy-3-chlorophenoxy)phenyl]acetate, octadecanoic acid, beta-amyrin, (2R,3S)-2-(3,4-dihydroxyphenyl)-3,4-dihydro-2H-chromene-3,5,7-triol, 3-dimethylamino-2-methyl-2-propinal, 12-octadecadienoic acid (Z,Z)-1-hexadecyne, scopolamine, propenoic acid-2-phenyl-,8-methyl-8-azabicyclo[3.2.1]octan-3-ylester, 8 azabicyclo[3.2.1]octane-3,6-diol, diacetate (ester), [1R-[1.alpha.,3.beta.(E),5.alpha.6.alpha.]]-, 2-butenoic acid, 2-methyl-,8-methyl-1-6-(1-oxopropoxy)-8-azabicyclo[3.2.1]oct-3-yl ester.
In general, the root extract contains a variety of major compounds, including phenolics, flavonoids, alkaloids, steroids, fatty acids, esters, hydrocarbons, terpenoids, aldehydes, and ketones. This study’s findings reveal a diverse array of components, such as fatty acid esters like 5-hexadecenoic methyl ester and octadecenoic acid methyl ester, carboxylic acids such as hexadecenoic acid and (Z,Z)-9,12-octadecenoic acid, and palmitic acid esters like hexadecenoic methyl ester.
Isolation of compounds from methanolic crude extract
In this study, column chromatography using silica gel (60–120 mesh) was employed for compound fractionation. A variety of gradient solvent systems ranging from 10:0 to 0:10 n-hexane/methanol ratios were utilized. A total of 22 fractions were gathered in a 50-mL volumetric flask from the initial extract, and the elution progress was tracked using thin-layer chromatography (TLC) on glass plates.
Throughout the investigation into thin-layer chromatography, various solvent systems were explored with the aim of achieving optimal resolution. Ultimately, it was determined that n-hexane: methanol ratio of 6:4 was the most suitable solvent system for analyzing the methanol extract obtained from D. fastuosa roots. Through the column chromatography fractionation process, a total of 21 fractions were obtained. Among these 21 fractions from the methanol extract, three fractions exhibited single spots. The purity of each fraction was evaluated using TLC plates, and their purity was further verified using melting point determination, which showed a sharp melting point range for one fraction, indicating purity, while the other two single-spot fractions showed a wider range of melting points, indicating lower purity. The respective Rf (Retention factor) values of these fractions were calculated and recorded.
The TLC plate confirmed the isolation of three compounds from the ranges of F3–F7, F9–F13, and F17–F21, which were labeled as the 1st (S-1), 2nd (S-2), and 3rd (S-3) compounds, with Rf values of 0.768, 0.779, and 0.846, respectively, indicating their potential purity. As previously mentioned, these compounds were isolated using a mixture of n-hexane and methanol (6:4) solvent ratios through gravity-assisted fractionation with low external pressure. Finally, 33 mg of S-1, 35 mg of S-2, and 13 mL of S-3 were obtained as potentially pure compounds from the methanol fractions. Subsequent melting point determination confirmed that S-2 had a sharp melting point, indicating purity, while the other two compounds displayed a relatively wider range of melting points, indicating lower purity. The compounds were then stored under suitable conditions (4 °C) for further analysis and characterization.
Melting point determination of isolated compounds
The melting points of compounds S1, S2, and S3 were assessed to determine their purity. The recorded melting point ranges were (225–228 °C), (230–231 °C), and (334–339 °C) respectively. Purity was determined by a melting point range not exceeding 2 °C. Consequently, S1 and S3 were deemed impure due to their broad melting point ranges of (334–339 °C) and (225–228 °C), respectively. Only compound S2 exhibited a narrow melting point range (230–231 °C). The melting point test revealed that S2 began melting at 230 °C and completely melted at 231 °C, indicating its purity. Conversely, the broad melting point ranges of S1 and S3 suggested impurities. Furthermore, the purity of the isolated S2 compound was confirmed through IR spectroscopy.
Structure analysis of isolated compound
Among the compounds that were isolated, the structure of the compound labeled as (S-2) was subjected to further structural analysis using various spectroscopic techniques, including UV spectroscopy, FT-IR spectroscopy, 1H-NMR spectroscopy, 13C-NMR spectroscopy, and DEPT-135. This analysis involved a comparison of the obtained results with reference data and information from the GC/MS analysis of the NIST Library.
UV–visible analysis
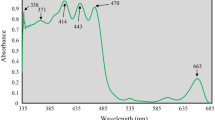
UV–Vis spectroscopic analysis was conducted to identify the phytoconstituents present in the extract. The qualitative UV–Vis spectrum profile of the methanol extract was chosen within the wavelength range of 300–800 nm due to the sharpness of the peaks and the presence of a well-defined baseline. This spectrum was generated to pinpoint compounds characterized by σ-bonds, π-bonds, lone pairs of electrons, chromophores, and aromatic rings.
The spectrum of the S-2 compound exhibited distinct bands at 371 nm and 408 nm with absorptions of 91.56 and 99.21, respectively. The UV–Vis spectrum obtained (figure not shown) illustrates that the absorption spectrum of methanol extracts from D. fastuosa roots is nearly transparent in the wavelength range of 300–800 nm. In the spectra, the presence of one or more peaks within the UV (MeOH) λmax (log ε) 200–410 nm range is a clear indicator of the existence of unsaturated groups and heteroatoms such as S, N, and O. In accordance with this, the spectrum of the D. fastuosa methanol extract displays two peaks at UV (MeOH) λmax (log ε) 371 (3.82) nm and 408 (3.92) nm. This confirms the presence of aromatic and other organic chromophores within the isolated compound.
FT-IR spectral analysis of compound S-2
The application of FT-IR spectroscopy is a crucial method for discerning the functional groups within active constituents found in a molecule. This is accomplished by assessing the molecule's response to energy absorption within the infrared spectrum. In the present investigation, FT-IR spectroscopy was employed to ascertain the existence of functional groups in the isolated pure compound, S-2, which was obtained from the methanol extract of D. fastuosa root. Subsequently, an analysis of the FT-IR spectrum was conducted to identify the functional group present in the compound. IR spectral interpretation faces challenges due to several factors. Residual solvents like methanol, natural lipids, waxes, polysaccharides, proteins, chlorophyll, and other pigments can all contribute additional peaks. Non-target plant materials, environmental contaminants, soil particles, and chemicals used in sample preparation can further complicate the spectra. Therefore, these factors were considered during spectral interpretation.
The FT-IR spectrum of S-2 shown in Fig. 2 provides valuable structural insights into the functional groups it contains. Specifically, the broad band at 3343 cm−1 is indicative of O–H stretching vibrations, suggesting the presence of a hydroxyl-related group, possibly an alcohol or phenol. In essence, this broad band at 3343 cm−1 corresponds to the hydroxyl group's O–H stretching, associated with non-hydrogen bonding alcohols or phenolic compounds, known for their antioxidant properties. Furthermore, the narrow, intense absorption band at 2927 cm−1 indicates the presence of –CH asymmetric stretching in an alkane, likely related to –CH3 stretching. Similarly, the narrow, weaker band at 2852 cm−1 is attributed to –CH asymmetric stretching in an alkane, potentially associated with –CH2– stretching. Additionally, a sharp band at 1710 cm−1 corresponds to the C=O stretching vibration, suggesting the presence of a carbonyl group within the methanol root extracts. The band at 1647 cm−1 exhibits weak intensity and is linked to C=C skeletal stretching in an alkene. Moreover, various overtone bands with variable intensities spanning from 1250 to 1600 cm−1 (specifically at 1517, 1467, 1376, and 1253 cm−1) indicate out-of-plane bending vibrations. Bands at 823 and 738 cm−1 suggest the availability of an ortho-para-disubstituted aromatic ring with alternative conjugate C=C functional groups. The weak band at 1050 cm−1 is associated with the C–O stretching vibration in alcohols.

FT-IR spectrum of S-2 compound from Datura fastuosa root extract
The FT-IR analysis also reveals the presence of phenolic and flavonoid functional groups, as evidenced by the O–H stretching, as well as phenols, alkaloids, flavonoids terpenes functional groups characterized by C–H asymmetric stretching. The distinctive characteristics of the bands in terms of their positions, shapes, and intensities enable the identification of fundamental components. The presence of C=O, C–H, C=C, C–O, and O–H bonds points to the presence of alkyl, alkene, alcohols, ethers, esters, carboxylic acids, anhydrides, and aromatic compounds which are typical of secondary plant metabolites (see Table 4). Therefore, the IR spectrum indicates the existence of carbonyl groups, alcohols, aromatics, as well as methylene and methyl groups attached to a quaternary carbon in the S-2 compound. Notably, there is no absorption observed in the region between 2220 and 2260 cm−1, which suggests the absence of a cyanide group in the isolated S-2 compound. This absence implies that the compound is free of cyanide and, consequently, non-toxic.
Proton nuclear magnetic resonance spectroscopy (1H-NMR)
1H-NMR spectroscopy is applied to characterize the hydrogen atom types in the isolated compound S-2 and to elucidate their connectivity (see Fig. 3). The 1H-NMR spectrum of the S-2 compound exhibits distinctive features, including resonances in the downfield chemical shift range, indicative of aromatic protons, connections involving olefinic double bonds on adjacent carbon atoms, and a notably congested up-field region owing to a higher concentration of aliphatic protons. Within the 1H-NMR spectroscopic data, a discernible OH group signal at δ 5.25 is observed. In the densely populated aliphatic region spanning δH 0.5–3.84, distinctive and characteristic signals associated with methyl protons has been observed.

1H-NMR spectrum of S-2 compound from Datura fastuosa root extract
The 1H-NMR spectrum of compound S-2 provided insights into the signal integrals and multiplicities. It revealed the presence of three aromatic protons in the range of δ [7.40–7.29] (m, 3H) and one non-aromatic proton at δ 5.25 (m-1H) adjacent to an electronegative atom. The detailed 1H NMR data (MeOD (Deuterated methanol), 500 MHz) included the following chemical shifts and multiplicity: δ 7.40–7.29 (4H, m, H-1), 7.37 (3H, dd, J = 8.0 Hz, H-2), 6.86—6.72 (2H, q, J = 9.5 Hz, H-3), 5.25 (1H, s, H-4), 4.17 (1H, d, J = 4.3 Hz, H-5), 4.11 (1H, tq, J = 9.7 Hz, H-6), 3.84 (1H, s, H-7), 3.63–3.41 (10H, m, H-8), 3.34 (1H, s, J = 2.34, H-8), 2.49 (1H, m, H-10), 2.82 (6H, s, H-11), 1.85 (3H, t, J = 7.3, H-12), 1.76 (3H, d, J = 6.2, H-13), 1.04 (9H, s, H-14), 0.99 (6H, s, H-15), totaling 49 protons in all.
In the 1H-NMR spectrum recorded at 500 MHz in MeOD, the signals included a range from δ 7.40–7.29 (m, 4H) for equivalent aromatic protons, a doublet of doublets at δ 7.37 (dd, J = 8.0 Hz, 3H) indicating aromatic protons, a singlet at δ 3.34 (s, 1H) for another aromatic proton, a quartet in the range of δ 6.86–6.72 (q, J = 9.5 Hz, 2H) representing a –CH2– group attached to methyl and carbonyl carbons, a singlet at δ 5.25 (s, 1H) corresponding to an aromatic –C–OH peak, a triplet quartet at δ 4.11 (tq, J = 9.7 Hz, 1H) suggestive of protons linked to adjacent carbon atoms and displaying a mixture of triplet and quartet characteristics due to interactions with both two equivalent (Hs) and three equivalent Hs, a peak at δ 3.84 (J = 5.3 Hz, 1H) representing protons attached to a tertiary carbon, peaks in the range of δ 3.63–3.41 (m, 10H) indicative of methylene protons, a singlet at δ 2.49 (s, 1H), and in the methyl range, nine peaks including two equivalent methyl singlets at δ 2.82 (s, 6H), signifying two equivalent –CH3 attachments in quaternary carbons of olefin functional groups, three equivalent methyl singlets at δ 1.04 (s, 9H) confirming the presence of three equivalent –CH3 attachments in quaternary carbons, one methyl triplet at δ 1.85 (t, J = 7.3 Hz), indicating one –CH3 attachment in a secondary carbon, and one methyl doublet at δ 1.76 (d, J = 6.2 Hz, 3H), suggesting one –CH3 attachment in a tertiary carbon, as well as two equivalent methyl singlets at δ 0.99 (s, 6H), indicating two equivalent –CH3 attachments in quaternary carbons. The 1H-NMR spectroscopy analysis of compound S-2 revealed the existence of a total of 49 proton signals distributed across various carbon environments, with nine –CH3, six –CH2–, four aromatic –CH–, five aliphatic –CH-, and one –OH group signal, each detected at distinct chemical shift regions as shown in Fig. 3.
Carbon-13 nuclear magnetic resonance spectroscopy (13C-NMR)
The compound S-2 was subjected to 13C-NMR analysis, which helped identify the types of carbons within the compound (see Fig. 4). The assignment of 13C-NMR data for S-2 was performed using both 13C-NMR and DEPT-135 spectra. The 13C-NMR spectrum of the isolated S-2 compound revealed a total of 32 distinct and well-resolved carbon signals. Among these signals, the most downfield peak corresponded to a ketone carbonyl carbon. These 32 carbon signals were categorized as follows: eight (8) methyl (sp3) carbons, five methylene (sp) carbons, two olefinic carbons, five methines, six aromatic ring carbons, four quaternary carbons, one keto carbonyl carbon, and one α-saturated keto carbonyl carbon. These assignments confirmed the presence of 32 non-equivalent carbon atom signals, aligning with the information derived from the DEPT-135 experiment. In the 400 MHz 13C-NMR spectrum of S-2 in MeOD, the downfield (deshield) region exhibited nine distinct signals with chemical shifts at δ [172.33, 136.28, 130.02, 129.90, 129.30, 128.49, 128.21, 115.96, and 115.57]. These were identified as one keto carbonyl carbon, six aromatic ring carbons, and two olefinic carbons. Specifically, the double-bonded carbons at δ 128.21 and 129.30 were attributed to the olefin carbons. Conversely, the up-field (shielded) region displayed 23 signals, including eight (8) methyl (–CH3) sp3 hybrid groups at δ [9.31, 19.89, 20.17, 22.95, 23.60, 24.25, 26.83], seven (7) methylene (–CH2–) groups at δ [20.17, 32.28, 34.90, 48.86, 49.00, 54.99, 57.71], six (6) methine (–CH–) groups at δ [29.97, 63.75, 70.36, 72.99, 73.47, 98.55], four (4) quaternary carbons at δ [29.68, 35.05, 65.42, 68.45], and one α-saturated keto carbonyl at δ 173.33. These assignments were supported by the NMR data detailed as follow.

13CNMR spectrum of S-2 compound from Datura fastuosa root extract
NMR (MeOD, 400 MHz) δ 172.33 (C-3, CO-Eth), 136.28 (C-32, C–OH), 130.02 (C, C-30), 129.90 (CH, C-34), 129.30 (C=C, C-20), 128.49 (C=C, C-18), 128.21 (C, C-30), 128.21 (CH, C-35), 115.96 (CH, C-33), 115.57 (CH, C-31), 98.55 (CH, C-4), 73.43 (CH, C-11), 72.99 (CH, C-15), 70.36 (CH, C-23), 68.45 (C, C-19), 65.42 (C, C-21), 63.77 (CH, C-29), 57.71 (CH2, C-25), 54.99 (CH2, C-9), 49.00 (CH2, C-28), 48.86 (α-CH2, CO-CH2-Me), 35.05 (C, C-10), 34.90 (CH2, C-13), 32.28 (CH2, C-8), 29.97 (CH, C-26), 29.68 (C, C-5), 26.63 (CH3, C-6), 26.63 (CH3, C-7), 24.25 (CH3, C-27), 23.60 (CH3, C-14), 22.96 (CH3, C-22), 20.17 (CH2, C-12), 20.17 (CH3, C-17), 19.89 (CH3, C-24), 9.81 (CH3, COCH2–CH3). Additionally, the ESIMS data indicated m/z 502 [M + H] +, and the positive ion HRESIMS data showed m/z 502.031 (calculated for C35H50O2 [M + H] +, 502.031), further corroborating the findings. In summary, the 13C-NMR results clearly revealed the presence of 32 non-equivalent carbon atoms in the S-2 compound, characterizing its structural composition.
Distortionless enhancement by polarization transfer (DEPT-135)
The S-2 compound's 1H NMR and 13C NMR assignments were determined through the DEPT-135 experiment, which distinguishes between carbon signals associated with different types of proton environments. In this experiment, –CH– and –CH3 groups produce positive signals, while –CH2– groups yield negative signals. The DEPT-135 spectrum displayed 16 signals corresponding to carbon atoms connected to odd protons (–CH– or –CH3) and six signals associated with carbon atoms bonded to even protons (–CH2–). This revealed the presence of a total of 23 distinct carbon atoms, with 17 linked to odd protons (δ values: 128.21, 115.96, 115.57, 98.55, 73.43, 72.99, 70.36, 63.77, 29.97, 26.63, 24.25, 23.60, 22.96, 20.17, 19.89, and 9.81) and seven connected to even protons (δ values: 57.71, 54.99, 49.00, 48.86, 34.9, 32.28, and 20.17) within the molecular structure (See Fig. 5).

DEPT-135 NMR spectrum of S-2 compound from Datura fastuosa root extract
The DEPT-135 spectrum displayed fewer signals than a standard 13C NMR spectrum because DEPT-135 exclusively detects carbons bonded to protons, omitting those without proton attachments. Carbon atoms linked to –OH groups (possibly hydroxyl) at δ 136.28, cyclohexane-linked aromatic carbons at δ 130.02 and 128.21, two olefinic carbons at δ 129.30 and 128.49, and a keto carbonyl carbon at δ 172.33 were all confirmed from the DEPT-135 spectrum. Furthermore, four peaks observed in the 13C NMR spectrum at δ values 29.68, 128.21, 68.45, and 65.42 were absent in the DEPT-135 spectrum, indicating that these carbons are classified as quaternary carbons. After a comprehensive analysis of the spectroscopic data, including 1H-NMR, 13C-NMR, DEPT-135, IR, and UV spectra for the S-2 compound isolated from the root of D. fastuosa and following a meticulous comparison with reference data and phytochemical compounds identified using GC/MS in the NIST Library (Search Libraries: D:\MassHunter\Library\NIST11.L), it was determined that the S-2 compound closely aligns with a phytochemical compound documented in the NIST Library. As a result, it is proposed and strongly suggested that the structure of compound S-2 is in good accordance with the structure provided in Fig. 6.

Proposed structure of S-2 compound from methanol root extract of Datura fastuosa
Discussion
The methanol root extract of D. fastuosa underwent phytochemical screening procedures and GC/MS analysis, revealing a diverse array of major compounds (see Tables 1 and 3). These include phenolics, flavonoids, alkaloids, steroids, fatty acids, esters, hydrocarbons, aldehydes, and ketones, most of which are known for their pharmacological properties. Previous studies have recognized their antioxidant, antibacterial, anti-inflammatory, anticancer, and antiviral effects [19, 20]. Additionally, the study unveiled specific components in the extract, such as fatty acid esters like hexadecenoic acid methyl ester and octadecenoic acid methyl ester, carboxylic acids like hexadecenoic acid and (Z,Z)-9,12-octadecenoic acid, and palmitic acid esters such as hexadecenoic acid methyl ester. These compounds exhibit a broad spectrum of bioactivities. Notably, compounds like octadecenoic acid methyl esters have previously been reported for their antibacterial, antifungal, and anticancer properties [21, 22]. Saturated fatty acids, including palmitic acid esters, have demonstrated a wide range of activities such as anticancer, antimicrobial, antioxidant, and anti-hemolytic effects [23]. Another significant compound, 2-methoxy-4-(2-propenyl) phenol, has been identified for its antibacterial, antimicrobial, antiseptic, anesthetic, and anticancer properties [24]. The presence of these bioactive compounds in the methanol root extract of D. fastuosa suggests its therapeutic potential in treating various ailments. The diverse range of identified compounds underscores the plant's pharmacological significance in traditional medicine. The UV–Vis spectroscopy analysis affirms the presence of aromatic and other organic chromophores within the isolated compound. However, it is essential to recognize the limitations of UV–Vis spectrophotometry when dealing with complex media. The challenge of attributing absorption peaks to specific constituents within the system is inherent. Thus, to ensure accurate characterization and identification of the constituents, findings from UV–Vis spectroscopy were augmented with other analytical techniques such as IR, GC/MS, and NMR. This comprehensive approach enhances the precision of characterizing the compound. Moreover, in addition to all the other functional groups detailed in the result section and depicted in Table 4, the FT-IR analysis of the S2 compound reveals the presence of phenolic and flavonoid functional groups, indicated by the O–H stretching, alongside phenols, alkaloids, flavonoids, and terpenes functional groups characterized by C–H asymmetric stretching. However, the accuracy of indicating terpenes through C–H asymmetric stretching is questionable since they were not detected in the phytochemical screening test of the methanol extract. Although other solvent extraction methods such as ethyl acetate and chloroform have suggested the presence of terpenoids in the root extract (data not included), It remains justifiable to deduce that the S2 compound is a phenolic compound and may well be a derivative of a diterpenoid.
The 1H-NMR, 13C-NMR, and DEPT-135 detection spectra of the S-2 compound exhibited a remarkable similarity, showing a 99% correspondence to the suggested compound structure in the NIST library. This structure is identified as 1-(7-(3-hydroxyphenyl)-1,1,4a,5,6,9,10a,10b-octamethyl-1,2,3,4,4a,4b,6a,7,8,9,10,10a,10b,11,12,12a-hexadecahydrochrysen-2-yl) propan-1-one, with a chemical formula of C35H50O2. The proposed structure likely represents a phenol and keto-containing bioactive compound. Therefore, based on the extensive spectroscopic data, chemical formula, and structural similarity, it is reasonable to conclude that the S2 compound aligns with the proposed structure in Fig. 6.
Conclusion
This investigation has successfully identified 49 potential bioactive chemical constituents within the methanolic root extract of D. fastuosa. Among these compounds, one has been isolated, purified and its structure elucidated. The presence of these bioactive substances in D. fastuosa reinforces its traditional medicinal use across diverse global communities, highlighting its potential significance in the development of novel drugs based on the structure of these isolated compounds. Datura fastuosa, with its array of bioactive constituents, shown as a plant of substantial phytopharmaceutical significance. Nonetheless, further research is imperative to assess the toxicity profile, biological activities, and pharmacological relevance of the isolated and identified phytochemical compounds.
Experimental
Plant sample collection, preparation, and extraction
Fresh roots of D. fastuosa were obtained from the Konso zone within the Southern Regional state of Ethiopia and the taxonomic verification of the plant was conducted. The freshly collected D. fastuosa roots were washed, rinsed with distilled water, and subsequently subjected to a 3-month period of shade drying in a well-ventilated laboratory. After drying, the plant material was finely ground to a particle size of 500 mill mesh using a combination of a local mortar and pestle as well as an electric blender, following the methodology outlined by Kiruthika and Sornaraj [25]. The resulting powdered sample was securely packed in aluminum foil and stored at room temperature in preparation for the extraction process. To initiate extraction, 140 g of dried D. fastuosa roots were submerged in 750 mL of colorless needles containing 99.97% MeOH. The soaking process extended over 72 h, during which periodic agitation occurred on an orbital shaker at 250 rotations per minute. Following the soaking period, the mixture was meticulously filtered using a Whatman #1 filter paper with a diameter of 320 mm and subsequently concentrated under reduced pressure using a rotary vacuum evaporator bath at 40 °C. The concentrated mixture was then preserved in a vial and stored in a labeled glass dish inside a refrigerator set at 4 °C until it was ready for further analysis, as outlined by Yaschilal et al. [26].
Phytochemical screening
The root extracts of D. fastuosa underwent a comprehensive phytochemical analysis following the established protocols outlined by Evans [27]. This analysis aimed to identify a spectrum of phytoconstituents, including alkaloids, phenolics, tannins, flavonoids, triterpenoids, terpenoids, saponins, and steroids within the plant material.
Gas chromatography–mass spectrometer (GC/MS) analysis
A 7820A gas chromatograph coupled to a 5977A inert mass detector from Agilent was employed for GC/MS analysis. This analytical technique was used to examine the phytochemical compounds responsible for various pharmacological activities in the given extract. The analysis was conducted on an Hp-5ms ultra-inlet capillary column (25 m × 250 µm and 0.25 µm). Helium gas with a purity of 99.999% served as the carrier gas, flowing at a rate of 1 mL min−1. The obtained analytical results were processed and interpreted using Mass Hunter Chem-Station. The separation process in the GC involved a temperature program starting at 60 °C for 0 min, it increased at a rate of 60 °C per minute to 240 °C, held for 1 min, and then increased at a rate of 100 °C per minute to 260 °C for 2 min. The GC oven was maintained at 290 °C, and the injection port temperature was held at 250 °C, with a solvent cut time of 2.50 min. The Mass Spectrometer program initiated at 3.00 min and concluded at 40.00 min, with an event time of 0.50 s and a scan speed of 1666 μL/s. All injections were made with a volume of 1 μL in splitless mode. The mass detector operated in full scan mode, covering the range m/z 50–550. The entire GC/MS analysis process took 40 min. The relative percentage of the extract was calculated by normalizing peak areas, following a previously reported method Aisha et al. [28].
Chromatographic methods
Column chromatography and thin-layer chromatography (TLC) were employed to isolate and purify bioactive molecules from the methanol root extract. The objective of this procedure was twofold: first, to determine the number of components present in the crude extract, and second, to assess the chemical proximity of these components. Additionally, it aimed to establish suitable solvent systems for column chromatography, as described by Popova et al. [29]. The methanol extract derived from D. fastuosa roots was applied to thin-layer chromatographic plates using a solvent system with an n-hexane to methanol ratio of 6:4.
Thin-layer chromatography (TLC) profile
A thin-layer chromatographic plate measuring 5 × 20 cm with a thickness of 0.5 mm was utilized, following the method described by Abdurohaman [2]. The extract was dissolved in methanol and manually applied to the plate using a capillary tube. The plate underwent development in n-hexane: methanol, employing varying trial ratios to increase the polarity of the solvent mixtures, including 10:0, 9:1, 8:2, 7:3, 6:4, 5:5, 4:6, 7:3, 8:2, 9:1, and 0:10, as outlined by Hemavathy et al. [30]. Furthermore, this method was applied to the sample using alternative mixed solvent systems, namely n-hexane:ethyl acetate:methanol:formic acid (4:3:2:1), n-hexane:ethyl acetate:acetic acid glacial (4:4:2), and n-hexane: methanol: chloroform (6:3:1), following the procedures as detailed by Ahamed et al. [31]. Subsequently, the spots were visualized under UV light at wavelengths of 254 nm and 365 nm. The solvent combination that displayed the best separation, i.e., n-hexane:methanol (6:4), was chosen for column chromatographic analysis. Data pertaining to the n-hexane:methanol (6:4) system was recorded on a datasheet by measuring the distance traveled by each individual spot and the distance covered by the solvent. Following visualization and labeling of the spots, the Rf was calculated using the formula: (Rf) = (Distance from the starting point to the center of the substance spot)/(Distance from the starting point to the solvent front). Consequently, the n-hexane:methanol (6:4) ratio was determined to be the appropriate column eluent for further analysis.
Column chromatography (CC)
The initial crude extract underwent fractionation via column chromatography using silica gel (60–120 mesh). In this process, a glass column with a cylindrical shape containing silica gel as the stationary phase was gradually filled from the top. A mobile phase consisting of n-hexane: methanol in a 6:4 polarity ratio flowed down the column under the influence of gravity and external pressure. The methanol extract, weighing 4 g, was absorbed into 20 g of silica gel and applied to the top of the column. The solvent was allowed to travel downward through the column, commencing with a combination of lower polarities and incrementally increasing their polarities, mirroring the TLC method. This approach enabled the compounds within the crude extract to interact differently with the stationary phase (silica gel) and the mobile phase, leading to their separation at distinct time intervals.
Consequently, the separation of compounds from the mixture was successfully accomplished, resulting in the collection of 22 individual fractions from the crude extract, all obtained using the n-hexane: methanol (6:4) ratio. The purity potential of each fraction was verified through TLC analysis. The individual compounds were subsequently analyzed and stored in a refrigerator at 4 °C for future structure elucidation, as described by Vivek et al. [32].
Structural elucidation of isolated compound
Spectroscopic methods were employed to establish the structure of the purified compound. The following techniques were utilized for this purpose: UV, FT-IR, 13C NMR, and 1H NMR spectroscopy.
UV–Vis spectroscopic analysis
A methanol-based solution containing an isolated sample at a concentration of 200 parts per million (ppm) was prepared for UV–Vis analysis. This initial sample was subsequently diluted at a 1:10 ratio using the same solvent. The resulting solution was then subjected to UV–Vis spectrophotometry, with a wavelength scan ranging from 300 to 800 nm to identify the λmax. During the analysis, characteristic peaks were identified, and the peak value of the UV–Vis spectrum was documented.
FT-IR spectroscopic analysis
Fourier-transform infrared spectroscopy (FT-IR) was employed to identify the distinctive functional groups within the D. fastuosa extract. A small quantity of the extract was blended with dry potassium bromide (KBr), and the resulting mixture was meticulously homogenized in a mortar. This blend was then subjected to a pressure of 6 bars within 2 min to create a thin KBr disc. Subsequently, the KBr disc was positioned in a sample cup designed for diffuse reflectance measurements. The IR spectrum was acquired using a Bruker Vertex 70 infrared spectrometer from Germany. The sample was scanned over a range from 4000 to 400 cm−1, and the peak values in the FT-IR spectrum were recorded.
Nuclear magnetic resonance (NMR) spectrophotometric analysis
The 13C NMR and 1H NMR spectra were acquired using a Bruker WM-400 UltraShield spectrometer, employing deuterated methanol (MeOD). The results are presented in δ ppm, utilizing tetramethylsilane (TMS) as the internal reference standard. In the 1H NMR spectra, splitting patterns were denoted as q for quartet, t for triplet, d for doublet, and s for singlet. Approximately 10 mg of the purified compound was dissolved in methanol (MeOD) and utilized for the analysis. The chemical shifts (δ) for both 1H NMR (MeOD, 500 MHz) and 13C NMR (MeOD, 100 MHz) spectra were recorded, and subsequent interpretation of the spectra was conducted.
DPPH free radical scavenging activity
To assess DPPH free radical scavenging activity, a 0.1 mM DPPH (2,2-diphenyl-1-picrylhydrazyl) solution was prepared by dissolving 4 mg of DPPH in 100 mL of methanol and stored in a dark bottle. Varying concentrations of plant extracts and ascorbic acid in methanol were prepared. In cuvettes, 200 µL of the DPPH solution and 20 µL of the plant extract or ascorbic acid were added, with 20 µL of methanol used for controls. The solutions were mixed well, incubated in the dark at room temperature for 30 min, and absorbance was measured at 517 nm using a UV–Vis spectrophotometer. The percentage inhibition was calculated using the formula Inhibition (%) = Acontrol − ASample/Acontrol × 100, where Acontrol is the absorbance of the DPPH solution with methanol and Asample is with the extract. The IC50 value was determined by plotting the inhibition percentage against extract concentrations.
Availability of data and materials
All data generated and analyzed during this study are included in this published article. Any additional information can be obtained from the corresponding author with reasonable request.
Abbreviations
- CC:
-
Column chromatography
- 13C-NMR:
-
Carbon-13 nuclear magnetic resonance spectroscopy
- D. fastuosa :
-
Datura fastuosa
- MeOD:
-
Deuterated methanol
- DEPT-135:
-
Distortionless enhancement by polarization transfer-135
- D. species:
-
Datura species
- FT-IR:
-
Fourier-transform infrared.
- 1H-NMR:
-
Proton nuclear magnetic resonance spectroscopy
- GC/MS:
-
Gas chromatography/mass spectrometry
- IUPAC:
-
International Union of Pure and Applied Chemistry
- NIST:
-
National Institute of Standards and Technology
- NMR:
-
Nuclear magnetic radiation spectrometry
- RDA:
-
Representational difference analysis
- Rf:
-
Retention factor
- SNK:
-
Student Newman Keuls
- TLC:
-
Thin-layer chromatography
- TMS:
-
Tetramethylsilane
- UV–Vis:
-
Ultra-violet visible spectrophotometry
- KBr:
-
Potassium bromide
- S-2:
-
Pure compound isolated from crude
- DPPH:
-
2-Diphenyl-1-picrylhydrazyl
References
Jamal P, Akbar I, Yumi Z, Irwandi J (2016) Process development for maximum lycopene production from selected fruit waste and its antioxidant and antiradical activity. J Food Process Technol 7:1–7. https://doi.org/10.5555/20163213263
Yessuf AM (2015) Phytochemical extraction and screening of bio active compounds from black cumin (Nigella sativa) seeds extract. Am J Life Sci 3:358–364. https://doi.org/10.11648/j.ajls.20150305.14
Pan W, Liu K, Guan Y, Tan GT, Hung NV, Cuong NM, Soejarto DD, Pezzuto JM, Fong HHS, Zhang H (2014) Bioactive compounds from Vitex leptobotrys. J Nat Prod 77:663–667
Ren Y, Benatrehina PA, Acuña UM, Yuan C, Chai HB, Ninh TN, De Blanco EJ, Soejarto DD, Kinghorn AD (2016) Isolation of bioactive rotenoids and isoflavonoids from the fruits of Millettia caerulea. Planta Med 82:1096–1104
Agarwal G, Wilson JR, Kurina SJ, Anaya-Eugenio GD, Ninh TN, Burdette JE, Soejarto DD, Cheng X, Carcache de Blanco EJ, Rakotondraibe LH, Kinghorn AD (2019) Structurally modified cyclopenta [b] benzofuran analogues isolated from Aglaia perviridis. J Nat Prod 82:2870–2877
Ren Y, De Blanco EJ, Fuchs JR, Soejarto DD, Burdette JE, Swanson SM, Kinghorn AD (2019) Potential anticancer agents characterized from selected tropical plants. J Nat Prod 82:657–679
Anaya-Eugenio GD, Addo EM, Ezzone N, Henkin JM, Ninh TN, Ren Y, Soejarto DD, Kinghorn AD, Carcache de Blanco EJ (2019) Caspase-dependent apoptosis in prostate cancer cells and zebrafish by corchorusoside C from Streptocaulon juventas. J Nat Prod 82:1645–1655
Ren Y, Elkington BG, Henkin JM, Sydara K, Kinghorn AD, Soejarto DD (2021) Bioactive small-molecule constituents of Lao plants. J Med Plants Res 15:540–559
Johari MA, Khong HY (2019) Total phenolic content and antioxidant and antibacterial activities of Pereskia bleo. Adv Pharmacol Sci 10:1–4
Veeresham C (2012) Natural products derived from plants as a source of drugs. J Adv Pharm Technol Res 3:200–201
Atanasov AG, Zotchev SB, Dirsch VM, The International Natural Product Sciences Taskforce, Supuran CT (2021) Natural products in drug discovery: advances and opportunities. Nat Rev Drug Discov 20:200–216
Raheem MAOA, Sulaiman FA, Abdulrahim HA, Ahmed O, Yusuf KO, Mukadam AA, Ganiyu LO, Kamilu LA, Oladepo KB, Alimi GO, Afolayan D, Odeniran T (2018) Assessment of environmental factors on secondary metabolites and toxicological effects of Datura metel leaves extracts. Ann Sci Technol 3:8–19. https://doi.org/10.2478/ast-2018-0015
Firdaus N, Viquar U, Kazmi MH (2020) Potential and pharmacological actions of Dhatura safed (Datura metel L.): as a deadly poison and as a drug: an overview. IJPSR 11:3123–3137. https://doi.org/10.13040/IJPSR.0975-8232.11(7).3123-37
Sharma M, Dhaliwal I, Rana K, Delta AK, Kaushik P (2021) Phytochemistry, pharmacology, and toxicology of Datura species—a review. Antioxidants 10:1291–1302
Al-Snafi AE (2022) The efficacy and safety of medicinal plants documented by clinical trials. World J Adv Pharm Med Res 3:30–77. https://doi.org/10.53346/wjapmr.2022.3.1.0036
Akharaiyi FC (2011) Antibacterial, phytochemical and antioxidant activities of Datura metel. Int J Pharm Technol Res 3:478–483
Donatus E, Ephraim C (2009) Isolation, characterization, and antibacterial activity of alkaloid from Datura metel Linn leaves. Afr J Pharm 3:277–281
Kokate K, Purohit A, Gokhale S (2008) Pharmacognosy, 42nd edn. Nirali Prakshan, Pune
Kumar R, Moorthy K, Vinodhini R, Punitha T (2013) Antimicrobial efficacy and phytochemical analysis of Indigofera trait Linn. Afr J Tradit Complement Altern Med 10:518–552
Saxena M, Saxena J, Nema R, Singh D, Gupta A (2013) Phytochemistry of medicinal plants. J Pharm Phytochem 1:168–182. https://doi.org/10.1007/978-1-4899-1778-2
Chandrasekaran M, Senthilkumar A, Venkatesalu V (2011) Antibacterial and antifungal efficacy of fatty acid methyl esters from the leaves of Sesuvium portulacastrum L. Med Pharmcol Sci 15:775–780
Cowan M (1999) Plant products as antimicrobial agents. Clin Microb Rev 12:564–582
Wei L, Wee W, Siong J (2011) Characterization of anticancer, antimicrobial, antioxidant properties and chemical compositions of Peperomia pellucida leaf extract. Acta Med Iran 49:670–674
Rao A, Zhang Y, Muend S (2010) Mechanism of antifungal activity of terpenoid phenols resembles calcium stress and inhibition of the tor pathway. Antimicrob Agent Chemother 54:506–529
Kiruthika K, Sornaraj R (2011) Screening of bioactive components of the flower Datura metel using the GC–MS technology. Int J Pharm Technol Res 3:2025–2028
Yaschilal M, Zewdu B, Eshetie M, Gedefaw G (2019) Evaluation of in vivo antidiabetic, antidyslipidemic, and in vitro antioxidant activities of hydromethanolic root extract of Datura stramonium L. (solanaceae). J Exp Pharm 11:29–38
Evans WC (2009) Chapter 17—General methods associated with the phytochemical investigation of herbal products. In: Evans WC (ed) Trease and evans pharmacognosy, 6th edn. Saunders Ltd, Philadelphia. https://doi.org/10.1016/B978-0-7020-2933-2.00017-4
Aisha M, Bashir L, Ndababru A, Abubakar A, Adisa M (2015) Phytochemical screening and GC–MS determination of bioactive constituents from methanol leaf extract of Senna occidentalis. J Coast Life Med 3:992–995
Popova I, Hall C, Kubátová A (2009) Determination of lignin’s in flaxseed using liquid chromatography with time-of-flight mass spectrometry. J Chromatogr 2:217–229
Hemavathy A, Shanthi P, Sowndharya C, Thiripura S, Priyadharshni K (2019) Extraction and isolation of bioactive compounds from a therapeutic medicinal plant—Wrightia tinctoria (Roxb.) R. Br. Int J Pharmacogn Phytochem Res 11:199–204
Ahamed T, Rahman M, Mohammad A (2017) Thin layer chromatographic profiling and phytochemical screening of six medicinal plants in Bangladesh. IJB 11:131–140
Vivek K, Bajpai L, Rajib M, Jae G (2016) Isolation and purification of plant secondary metabolites using column-chromatographic technique. J Pharmcol 11:844–848
Acknowledgements
The authors express gratitude for the support and facilities from their respective universities. Special thanks are also extended to Mr. Teykanto Kasso (Urkupile), a traditional medicine healer, for generously sharing profound knowledge about the plant and its traditional uses. Taxonomic Verification of D. fastuosa Roots: Adherence to Local, National, and International Guidelines: Datura fastuosa roots were collected from the Konso zone in the Southern Regional State of Ethiopia. The plant underwent taxonomic verification at the Biology/Botany department of Arba Minch University College of Natural and Computational Sciences, situated in Arba Minch, Ethiopia, adhering to local, national, and international guidelines.
Funding
The authors did not receive support from any organization for the submitted work.
Author information
Authors and Affiliations
Contributions
Girma Mengesha Melese performed the experimental work, conducted data analysis, interpreted the results, and authored the initial manuscript. Matthewos Eshete made contributions to the project in terms of design, reviewing, as well as the analysis and interpretation of the results. Tewodros Brihanu Aychiluhim played a significant role in providing guidance for result interpretation, reviewing, and offering overall research support. Abdurrahman Mengesha Yessuf actively participated by providing advice for result interpretation, contributing to the reviewing process, and offering support for the overall research endeavor.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This article does not involve any studies conducted on human participants or animals by any of the authors.
Consent for publication
All authors involved in the preparation of this manuscript have thoroughly reviewed and approved its content for submission.
Competing of interests
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Melese, G.M., Aychiluhim, T.B., Yessuf, A.M. et al. Identification and characterization of phytochemicals in methanolic extract of roots of Datura fastuosa using various techniques. Futur J Pharm Sci 10, 108 (2024). https://doi.org/10.1186/s43094-024-00682-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43094-024-00682-6