
Abstract
Background
In contemporary society, anxiety has become a widespread disorder leading to compromised well-being and heightened depressive states. Extensive literature reviews indicate the diverse biological effects of benzimidazole and piperazine derivatives, notably their impact on the central nervous system. This study aimed to design, molecularly dock, synthesize, and assess the anxiolytic potential of six derivatives of 2-(4-phenylpiperazin-1-yl)-1H-benz[d]imidazole and 2-(4-phenylpiperazin-1-methyl)-1H-benz[d]imidazole.
Results
In the present study, an attempt was made to synthesize benzimidazole derivatives conventionally. The benzimidazole nuclei are condensed with various substituted piperazines to obtain targeted benzimidazole–piperazine hybrids. Their anxiolytic activity is determined using the Elevated Plus Maze test and hole board test in mice. All compounds have shown good docking scores and in vivo anxiolytic activity.
Conclusion
Out of all the derivatives synthesized, compounds 5b, 5c, and 5f exhibited outstanding anxiolytic efficacy in both computational simulations and live subjects. Compound 5b demonstrated a remarkable docking score relative to the ligand, suggesting its potential as a promising candidate warranting further exploration.
Similar content being viewed by others


Background
Anxiolytics, crucial for managing anxiety and enhancing mental well-being, should be used under medical supervision to mitigate symptoms of anxiety disorders and enhance overall health and quality of life [1]. These medications slow down the central nervous system (CNS), primarily by enhancing the activity of gamma amino butyric acid (GABA), a neurotransmitter that induces CNS depression. This class of drugs, known for their calming effects, encompasses benzodiazepines, ethanol, opiates, and barbiturates [2, 3]. Benzodiazepines, for instance, potentiate GABA neurotransmission by increasing the affinity of GABA receptors, thereby intensifying inhibitory effects on the CNS [4, 5]. Additionally, heterocyclic compounds like benzimidazole and piperazine, noted for their diverse pharmacological properties including antibacterial, antiviral, antidiabetic, and anticancer activities, have garnered attention for potential drug development [6,7,8,9,10,11]. The current study synthesizes novel benzimidazole–piperazine derivatives and evaluates their anxiolytic activity.
Materials
The docking simulations were conducted using an Asus personal computer. Autodock Vina software was utilized for docking studies, while Swiss ADME and Molinspiration software were employed to predict the pharmacokinetic properties of the compounds. PASS online software was utilized to forecast the biological activity of the designed compounds. GraphPad Prism 5 software was employed for statistical analysis of biological activity data.
Infrared spectra were acquired using an ATR (Attenuated Total Reflectance) spectrophotometer (Bruker). Proton resonance magnetic spectra (1H NMR) were recorded at 400 MHz, and chemical shifts were expressed in “δ ppm”. Mass spectra were obtained from SPPU, Pune, with molecular peaks expressed in the m/z ratio. Thin-layer chromatography was employed for reaction monitoring using an iodine chamber.
Methods
Docking
The crystal structure of the Human GABA-A receptor alpha1-beta2-gamma2 subtype complexed with GABA and flumazenil, conformation B (PDB ID: 6D6T) [12], was obtained from the Protein Data Bank in PDB format. This structure underwent cleaning procedures, involving the removal of water molecules, co-crystallized ligands, and non-essential entities, using Discovery Studio Visualizer. Subsequently, the cleaned structure was converted to pdbqt format after appropriate charge assignment. Ligand structures were designed using Marvin Sketch version 5.8.1, Chem Axon. Both protein and ligand structures were prepared utilizing Auto Dock Tools following a previously established protocol [13]. A search space grid was generated around the binding site of the GABA receptor using Auto Dock Tools. The grid dimensions were adjusted to encompass the active site to ensure a comprehensive exploration of ligand conformations. Auto Dock Vina was used to carry out docking between the prepared ligands and proteins. The conformation possessing the least binding energy was further analyzed to study its binding mode with the receptor. The protein–ligand interactions were viewed using BIOVIA, Dassault Systems, Discovery Studio 16.0.1, San Diego: Dassault Systems, 2016.
Chemistry
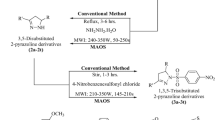
A series of novel piperazine-linked benzimidazole analogs (5a–f) were synthesized via a three-step synthetic pathway outlined in Fig. 1. Initially, 2-oxo benzimidazole (1) was synthesized by condensing o-phenylene diamine (OPD) and urea at 135–400 °C in DMF. Subsequently, halogenation was achieved by adding POCl3 and phenol crystals, allowing the reaction to proceed at 104–107 °C to yield 2-chlorobenzimidazole (2), serving as a core structure for derivatives [14,15,16]. Another core structure, 2-chloromethyl benzimidazole (3), was synthesized by refluxing O-phenylene diamine and chloroacetic acid with 5N HCl [17, 18]. The piperazine substituents (4a–d) were obtained by refluxing Bis-2-chloroethylamine hydrochloride with substituted anilines at 142–145 °C in the presence of p-toluene sulphonic acid (PTSA) using xylene as a solvent [19, 20]. Lastly, equimolar quantities (0.009 mol) of the core structure and substituted piperazines were dissolved separately in 1,4-dioxane, mixed, and refluxed with triethylamine (TEA) as a catalyst for 12 h [21]. The resulting precipitate was collected using chilled water, filtered by suction, and recrystallized from ethanol after drying to obtain the target derivatives (5a–f).

Scheme of synthesis of benzimidazole derivatives
All synthesized analogs underwent characterization via infrared spectroscopy (IR), proton nuclear magnetic resonance spectroscopy (1H NMR), and mass spectrometry (MS) for molecular weight determination. IR spectra revealed characteristic bands at 3300–3400 cm−1 corresponding to the Aromatic N–H bond, 900–1100 cm−1 for the C–F stretch, and 2800 cm−1 for the piperazinyl aliphatic C–H bond [22, 23]. In the 1H NMR spectra, the distinctive singlet peak of the benzimidazole nucleus was observed between 12.5 and 13.6 ppm for all derivatives. Peaks corresponding to piperazinyl aliphatic C–H appeared at 3.5–4.2 ppm, aromatic C-H peaks were detected at 7–8 ppm, and methylene C-H peaks for nucleus (3) were observed at 3.8–4.0 ppm [22, 23]. Mass spectrometry results corroborated the molecular weights of the compounds, with an additional weight corresponding to proton addition.
Biological evaluation
The anxiolytic activity was assessed using the Hole Board Test and Elevated Plus Maze (EPM) test in Swiss albino mice, with diazepam employed as the standard reference [24, 26]. The Hole Board Test apparatus comprised a wooden floor measuring 40 cm × 40 cm, elevated 2 inches above the ground, featuring sixteen holes arranged symmetrically in a diamond pattern. Groups of 5 animals were individually placed on one edge of the apparatus and monitored for 5 min to record the number of pockings. Diazepam (4 mg/kg i.p) was administered 30 min before the test.
For the EPM test, the wooden apparatus was elevated to a minimum height of 50 cm from the ground, adhering to the specifications described by Lister [25, 26]. The EPM consisted of two open arms (50 × 10 cm) and two closed arms (50 × 10 × 40 cm). Each group of 5 animals was individually positioned in the center of the EPM, facing the closed arms, and their time spent in both open and closed arms was recorded over 5 min.
Results
Synthesis and spectral data
2-(4-(4-Fluorophenyl)piperazin-1-yl)-1H-benz[d]imidazole (5a)
Yield: 56%; m.p: 216–218 °C; IR Ranges (ATR, cm−1); N–H stretch: 3337.25, C–H aromatic: 3041.76, C–H aliphatic: 2813.17, C–H aliphatic bend (CH2):1433.00, C=C stretch: 1600.32, C–N aromatic: 1210.90, C–F stretch: 999.71. H1 NMR Shifts; Aromatic C–H: (m 4H): 7.04–7.20, Piperazine C–H: (m 8H): 3.16–3.50, Aromatic C–H: (m 4H): 7.63–7.72, Benzimidazole N–H: (s 1H): 13.04. MASS: MOLECULAR ION PEAK (297.1523).
2-((4-(4-Fluorophenyl)piperazin-1-yl)methyl)-1H-benz[d]imidazole (5b)
Yield: 60%; m.p: 159–163 °C; IR Ranges (ATR, cm−1); N–H stretch: 3491.91, C–H aromatic: 3025.29, C–H aliphatic: 2908.47, C–H aliphatic bend (CH2): 1475, C=C stretch: 1650.50, C–N aromatic: 1204.77, C–F stretch: 1011.75. H1 NMR Shifts; Aromatic C–H: (m 4H): 7.16–7.21, Piperazine C-H: (m 8H): 3.29–3.88, Aromatic C–H: (m 4H): 7.44–7.63, methylene C–H: (s 2H): 3.89, Benzimidazole N–H: (s 1H): 13.61. MASS: MOLECULAR ION PEAK (311.1670).
2-((4-(3-Fluorophenyl)piperazin-1-yl)methyl)-1H-benz[d]imidazole (5c)
Yield: 70%; m.p: 122–126 °C; IR Ranges (ATR, cm−1); N–H stretch: 3337.25, C–H aromatic: 3041.55, C–H aliphatic: 2813.17, C–H aliphatic bend (CH2): 1433.00, C=C stretch: 1600.32, C–N aromatic: 1210.90, C–F stretch: 999.71. H1 NMR Shifts; Aromatic C–H: (m 4H): 7.05–7.17, Piperazine C–H: (m 8H): 3.39–3.88, Aromatic C–H: (m 4H): 7.17–7.44, methylene C–H: (s 2H): 3.89, Benzimidazole N–H: (s 1H): 13.61. MASS: MOLECULAR ION PEAK (311.1660).
2-((4-(2-Fluorophenyl)piperazin-1-yl)methyl)-1H-benz[d]imidazole (5d)
Yield: 71%; m.p: 116–119 °C; IR Ranges (ATR, cm−1); N–H stretch: 3481.78, C–H aromatic: 3038.96, C–H aliphatic: 2809.86, C–H aliphatic bend (CH2): 1433.42, C=C stretch: 1601.73, C–N aromatic: 1233.37, C–F stretch: 1016.61. H1 NMR Shifts; Aromatic C–H: (m 4H): 6.99–7.17, Piperazine C–H: (m 8H): 3.30–3.50, Aromatic C–H: (m 4H): 7.17–7.61, methylene C–H: (s 2H): 3.89, Benzimidazole N–H: (s 1H): 13.61. MASS: MOLECULAR ION PEAK (311.1675).
2-((4-Phenylpiperazin-1-yl)methyl)-1H-benz[d]imidazole (5e)
Yield: 57%; m.p: 162–165 °C; IR Ranges (ATR, cm−1); N–H stretch: 3481.78, C–H aromatic: 3038.96, C–H aliphatic: 2809.86, C–H aliphatic bend (CH2): 1433.42, C=C stretch: 1601.73, C–N aromatic: 1233.37. H1 NMR Shifts; Aromatic C–H: (s 1H, m 3H): 6.99–7.20, Piperazine C–H: (m 8H): 3.19–3.46, Aromatic C–H: (m 4H): 7.50–7.57, methylene C–H: (s 2H): 4.07, Benzimidazole N–H: (s 1H): 12.50. MASS: MOLECULAR ION PEAK (280.1690).
2-(4-(3-Fluorophenyl)piperazin-1-yl)-1H-benz[d]imidazole (5f)
Yield: 64%; m.p: 195–199 °C; IR Ranges (ATR, cm−1); N–H stretch: 3337.25, C–H aromatic: 3041.55, C–H aliphatic: 2813.17, C=C stretch: 1600.32, C–N aromatic: 1210.90, C–F stretch: 999.71. H1 NMR Shifts; Aromatic C–H: (m 4H): 7.07–7.20, Piperazine C–H: (m 8H): 3.16–3.50, Aromatic C–H: (m 4H): 7.20–7.63, Benzimidazole N–H: (s 1H): 13.04. MASS: MOLECULAR ION PEAK (297.1505).
The thin layer chromatography (TLC) characterization data for the synthesized substituents and derivatives, as well as spectra for infra-red (IR), proton nuclear magnetic resonance (1H NMR), and Mass analyses of the synthesized derivatives, are provided in the Additional files 1 and 2.
Biological activity (anti-anxiety activity)
The anxiolytic activity of the compounds was assessed through two tests: the Hole Board Test and the EPM Test. The experimental protocol for both tests was as follows: Group I received vehicle treatment (0.5% carboxymethyl cellulose in water). At the same time, Group VIII served as the standard reference with diazepam administration at 4 mg/kg intraperitoneally (i.p). Groups II to VII received test compounds 5a–f (synthesized compounds) at 50 mg/kg orally (p.o). Comparisons were made between Groups II to VIII and Group I, and the results were subjected to statistical analysis using GraphPad Prism 5 software, employing one-way ANOVA followed by Dunnett’s test.
Hole-board test
Each of the eight groups, consisting of five mice per group, was individually monitored for the number and duration of pocking in the hole board apparatus over 5 min. A higher number and longer pocking duration indicate the compound's anti-anxiety properties being evaluated. The mean ± standard error of the mean (SEM) for each group is presented in Table 1.
Compounds within Groups III, IV, and VII (Derivatives 5b, 5c, and 5f) exhibited very highly significant anxiolytic activity (***p < 0.001) in comparison to the control group, as assessed by the number of pocking. Compounds across Group II-VII (Derivatives 5a–f) demonstrated highly significant anxiolytic activity relative to the control group, as determined by the duration of pocking.
Figure 2 presents a graphical depiction of the data obtained from the hole board test. It is evident from the graph that all derivatives exhibit significant anxiolytic effects compared to both the control and standard groups.

Graphical representation of anxiolytic activity by Hole board apparatus. (A Number of pocking; B duration of pocking, *significant, **highly significant, ***very highly significant by ANOVA and Dunnett’s test)
EPM test
T Each of the eight groups, comprising five mice per group, was individually monitored for the number of entries and the duration of entries into the open arms of the EPM apparatus over a 5-min duration. An increased number and duration of entries into the open arms of the EPM are indicative of the anti-anxiety properties of the compound being evaluated. The mean ± standard error of the mean (SEM) for each group is presented in Table 2.
Discussion
Using Molinspiration [27] and Swiss ADME software [28], the physicochemical parameters and ADME (absorption, distribution, metabolism, and excretion) properties of all compounds were comprehensively examined before synthesis. Lipinski's rule of five (RO5), which evaluates essential physicochemical properties crucial for a molecule's efficacy, safety, or metabolism, was considered for all compounds.
Ideally, oral drugs should possess a Log P value < 5, a molecular weight < 500, a topological polar surface area (TPSA) < 90 to efficiently cross the blood–brain barrier, a molar refractivity value between 40 and 130, fewer than 10 hydrogen bond acceptors, and fewer than 5 hydrogen bond donors [29]. The assessment revealed that none of the compounds violated Lipinski's rule of five (RO5), as indicated in Table 3. This suggests that all derivatives were well-designed and exhibit desirable drug-like or pharmacological characteristics, rendering them potentially accessible for oral administration.
The blood–brain barrier (BBB) penetration, permeability glycoprotein (Pgp) substrate, gastrointestinal (GI) absorption, and cytochrome P450 (CYP450) enzymes, particularly CYP2C19 inhibitors, are key pharmacokinetic properties indicative of favorable drug likeliness potential. Utilizing Swiss ADME software, an in-silico assessment of pharmacokinetic properties was conducted, as outlined in Table 4.
All compounds examined in the ADME software exhibited high gastrointestinal absorption. Furthermore, in the ADME study, all derivatives were identified as Pgp (glycoprotein pump) substrates. P-glycoprotein plays a significant role in restricting the cellular uptake of medications from the bloodstream into the brain and from the intestinal lumen into epithelial cells. While positive Pgp substrate properties can diminish medication absorption, this effect can be mitigated by increasing the dosage, as high drug concentrations in the intestinal lumen can saturate P-glycoprotein transport function.
All derivatives are expected to inhibit the liver enzyme CYP3A4, while CYP2C19 remains unaffected. Consequently, the results indicate that the synthesized derivatives possess favorable pharmacokinetic and physicochemical properties, suggesting their potential utility as effective lead compounds with notable membrane permeability and oral bioavailability
An initial evaluation of central nervous system (CNS) depressant activity was conducted using the PASS study [30]. The derivatives exhibited virtually significant anxiolytic and antipsychotic activity. Values above 0.2 are considered significant according to PASS evaluation criteria, as depicted in Table 5.
All derivatives underwent a docking study to assess their interaction with the active site of the inhibitory ion channel Human GABA-A receptor alpha1-beta2-gamma2 subtype complexed with GABA and flumazenil, conformation B (PDB ID 6D6T), aiming to determine binding energy and interactions. It was observed that all derivatives exhibited favorable binding energy and engaged in various interactions with amino acids at the active site. The reference compound, flumazenil, exhibited substantial affinity towards the receptor molecule via halogen bonding with HIS D:102; hydrogen bonding to THR E:142; and van der Waals interactions with SER D:206, ASP E:56, and MET E:130; pi-pi stacking interactions with PHE E:77, TYR E:58, TYR D:160, and TYR D:210 in addition to other bonding modalities, accompanied by the binding energy of − 9.4 kcal/mol. The synthesized compounds exhibited binding energies ranging from − 8.4 to − 9.5. Details of the docking study outcomes are presented in Table 6. The derivatives 5a, 5b, 5c, and 5e exhibit hydrogen bonding interactions with the protein, as illustrated in Fig. 3. The principal binding interactions vis-a-vis the ligand flumazenil are deliberated upon herein. Compound 5a forms a single hydrogen bond with the active site residue SER D:206, in addition to engaging in two pi-pi stacking interactions with TYR E:58 and PHE E:77. Compound 5b establishes two hydrogen bonds with active site residues HIS D:102 and SER D:206, along with pi-pi stacking interactions with TYR D:160 and TYR D:210. Similarly, compound 5c forms two hydrogen bonds with SER D:206 and demonstrates halogen (fluorine) interaction with SER D:159, as well as pi-pi stacking with TYR D:210. Compound 5e forms a lone hydrogen bond with SER D:206. Besides these hydrogen bonds, pi-pi stacking interactions with residues TYR D:160 and TYR D:210, along with pi-anion interactions with ASP E:56 and MET E:130, appear to significantly contribute to the molecule's binding to the receptor. In contrast, Compounds 5d and 5f do not establish any hydrogen bonds with the receptor.

2D and 3D docking interactions of derivatives 5a, 5b, 5c and 5e
The anxiolytic efficacy of these compounds was evaluated in comparison to a control group. In the hole board test, control group mice exhibited fewer instances of pocking indicative of fear and anxiety, whereas the treated groups displayed an increased number and duration of pocking due to the anxiolytic effects. Compounds 5b, 5c, and 5f demonstrated notably significant anxiolytic activity based on the frequency of pocking, while all compounds 5a-f exhibited significant activity in terms of pocking duration. During the EPM test, rats administered with anxiolytic drugs spent more time in the open arm compared to the control group, reflecting heightened anxiety in the animals on an elevated platform. Notably, all six synthesized derivatives (5a–f) displayed highly significant anxiolytic activity in the EPM test.
Conclusion
All newly synthesized benzimidazole derivatives 5a–f have exhibited highly significant biological anti-anxiety activity. Most compounds have demonstrated drug-like properties, along with favorable gastrointestinal absorption and bioavailability as predicted by virtual computational tools. These findings are encouraging and warrant further investigation.
Moreover, docking studies against the Human GABA A receptor (PDB ID: 6D6T) revealed favorable binding energies for all derivatives, validating our hypothesis regarding the potential of the recently synthesized 2-(4-phenylpiperazin-1-yl)-1H-benz[d]imidazole and 2-((4-phenylpiperazin-1-yl) methyl)-1H-benz[d]imidazole derivatives as lead candidates for anti-anxiety drug development. Notably, compound 5b exhibited superior binding affinity compared to the ligand and demonstrated excellent in vivo anxiolytic activity, thus presenting a promising candidate for further exploration.
Availability of data and materials
The data that support the findings of this study are available from the corresponding author, upon reasonable request.
Abbreviations
- ADME:
-
Absorption distribution metabolism elimination
- ANOVA:
-
Analysis of variance
- ASP:
-
Aspartic acid
- BBB:
-
Blood–brain barrier
- CPCSEA:
-
Committee for Control and Supervision of Experimental on Animals
- CYP450:
-
Cytochrome P450
- EPM:
-
Elevated Plus maze
- GABA:
-
Gamma amino butyric acid
- GI:
-
Gastro-intestinal
- GPCR:
-
G-protein coupled receptor
- HIS:
-
Histidine
- IAEC:
-
Institutional Animal Ethics Committee
- IR:
-
Infra-red
- MET:
-
Methionine
- NMR:
-
Nuclear magnetic resonance
- PASS:
-
Prediction of activity spectra for substance
- Pgp:
-
Permeability glycoprotein
- PHE:
-
Phenylalanine
- PTSA:
-
Para toluene sulfonic acid
- SEM:
-
Standard error of mean
- SER:
-
Serine
- TLC:
-
Thin layer chromatography
- TPSA:
-
Topological polar surface area
- TYR:
-
Tyrosine
References
Bistrisky A, Khalsa S, Cameron M, Sciffman J (2013) Current diagnosis and treatment of anxiety disorders. Pharm Ther 38(1):30–38 (PMID: 23599668; PMCID: PMC3628173)
Rang HP, Dale NM, Riter JM, Flower RJ (2003) Rang and dells pharmacology. Elsevier
Tripathi KD (1998) Essential of medicinal pharmacology. Jaypee Brothers Medical Publications, New Delhi
Remers J, Wilson W, Gisvold (2002) Textbook of organic medicinal and pharmaceutical chemistry. Lippincott Williams and Wilkins, Philadelphia
Salguirao JB, Ardenghi P (1997) Anxiolytic natural and synthetic flavonoid ligands of the central benzodiazepine receptor have no effects on memory tasks in rats. Pharmacol Biochem Behav 54(4):887–891. https://doi.org/10.1016/s0091-3057(97)00054-3
Foye WO, Lamke MT, Williams DA (1995) Principles of medicinal chemistry. B.I. Waverly, New Delhi
Barar FS (2000) Essentials of pharmacotherapeutics. S. Chand and Company Ltd, New Delhi
Venkatramana C, Singh A, Tiwari A, Tiwari V (2010) Synthesis of phenyl hydrazine substituted benzimidazole derivatives and their biological activity. IJPSR 1:34–38. https://doi.org/10.13040/IJPSR.0975-8232.1(1).34-38
Kumar D, Bhat Z, Kumar V, Shah M (2011) Nature: anxiolytics in the lap of nature. Webmedcentral Pharm Sci. https://doi.org/10.13140/RG.2.2.25524.55684
Walia R, Hedietullah MD (2011) Benzimidazole derivatives: an overview. IJRPC 1:3565–3574
Chioua R, Benabdelouahab F, Chioua M, Martínez R (2002) Piperazine. Molecules 7(2002):507–510. https://doi.org/10.3390/70700507
Human GABA-A receptor alpha1-beta2-gamma2 subtype in complex with GABA and flumazenil, conformation B (PDB ID: 6D6T). https://www.rcsb.org/structure/6d6t
Unavane SA, Syed S, Jain HK, Bansode A (2023) Docking and pharmacokinetic studies for screening terpenoids from erythroxylum species as anticancer agents. Lett Drug Des Discov 20(12):2025–2033. https://doi.org/10.2174/1570180819666220929121630
Yekkirala V, Sudhagani RK, Panuganti L (2013) Facile and efficient one-pot synthesis of benzimidazoles using lanthanum chloride. Org Med Chem Lett 3:1–8. https://doi.org/10.1186/2191-2858-3-7
Kumar K, Pathak DP (2012) Synthesis characterization and evaluation of 2-substituted benzimidazole derivative. Pharma Innov 1:44–50
Dubey PK, Naidu A, Anandam V (2005) Synthesis of 1- (n-hexyl-5-one)-2 chlorobenzimidazole. Ind J Chem 44(B):1239–1242
Petkar K, Parekh P, Mehta P (2013) Synthesis and evaluation of 2-chloromethyl-1h-benzimidazole derivatives as antifungal agents. Int J Pharm Pharm Sci 5:115–119
Conrad M, Assman L, Wroblowsky H, Casser C, Bielefeldt D (2000) Process for preparing 2-chlorobenzimidazole derivatives. US Patent 6020200A, 22 Feb 2000
Pai R, Diptanshu A (2012) Synthesis of new analysis 3,4-dihydro-quinazolinone derivatives as a typical antidepressant, sedative, and antiparkinsons agents. J Heter Lett 2:117–128
Jain VK, Jain B, Sharma K, Saha D (2011) Synthesis characterization and antimicrobial screening of some 4-substituted 1(4substituted phenyl) piperazine derivatives. Int J Curr Pharm Res 3:66–70
Malik S, Ahuja P (2014) Design and synthesis of new of 3-(benzo[d]isoxazol-3-yl)-1-substituted pyrrolidine-2, 5-dione derivatives as anticonvulsants. Euro J Med Chem 84:42–50. https://doi.org/10.1016/j.ejmech.2014.07.016
Pavia DL, Lampman GM, Kirz GS (2007) Introduction to spectroscopy. Thomson Learning Publications
Xia Z, Wei Q, Yang Q, Qiao C (2012) Lanthanide coordination compounds with 1H-benzimidazole-2-carboxylic acid: syntheses, structures and spectroscopic properties. CrystEngComm 15:86–99. https://doi.org/10.1039/C2CE26120K
Barua CC, Roy JD (2009) Anxiolytic effect of hydroethanolic extract of Drymaria cordata L Willd. Ind J Exp Biol 47:969–973 (PMID: 20329700)
Czopek A, Byrtus H (2010) Synthesis and pharmacological evaluation of new 5-(cyclo) alkyl-5-phenyl- and 5-spiroimidazolidine-2,4-dione derivatives. Novel 5-HT1A receptor agonist with potential antidepressant and anxiolytic activity. Euro J Med Chem 45:1295–1303. https://doi.org/10.1016/j.ejmech.2009.11.053. (PMID: 20060623)
Nannapaneni DT, Gupta A, Reddy MI (2010) Synthesis, characterization, and biological evaluation of Benzimidazole derivatives as potential anxiolytics. J Young Pharm 2:273–279. https://doi.org/10.4103/0975-1483.66809. (PMCID: PMC2964780)
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (1997) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 23:4–25. https://doi.org/10.1016/s0169-409x(00)00129-0. (PMID: 11259830)
Acknowledgements
Authors thank Savitribai Phule Pune University for providing the spectral data.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
All authors have read and approved the manuscript. BM: synthesis, characterization, in silico evaluation, and outline of study; LK: design of compounds, synthetic scheme; VN: animal model; SU: docking studies; LS and PK: biological activity work.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
IAEC/Dec2014/06.
Consent for publication
The authors declare no conflict of interest.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
: Spectral data.
Additional file 2
: Thin Layer Chromatography data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mahajan, B.S., Kawale, L.A., Nade, V.S. et al. Design, synthesis, and evaluation of anxiolytic activity of 2-(4-phenylpiperazin-1-yl)-1H-benz[d]imidazole and 2-(4-phenylpiperazin-1-methyl)-1H-benz[d]imidazole derivatives. Futur J Pharm Sci 10, 50 (2024). https://doi.org/10.1186/s43094-024-00626-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43094-024-00626-0