
Abstract
Background
The Junín virus (JUNV) is well known for causing argentine haemorrhagic fever (AHF), a severe endemic disease in farming premises. The glycoprotein of JUNV is an important therapeutic target in vaccine design. Despite using drugs and neutralizing weakened antibodies being used in the medication, neither the severity reduced nor eradicated the infection. However, this constraint can be resolved by immunoinformatic approaches.
Results
The glycoprotein fasta sequence was retrieved from NCBI to anticipate the B cell and T cell epitopes through the Immune Epitope Database. Furthermore, each epitope underwent validation in Vaxijen 2.0, Aller Top, and Toxin Pred to find antigenic, nonallergic, and non-toxic peptides. Moreover, the vaccine is designed with appropriate adjuvants and linkers. Subsequently, physicochemical properties were determined in ProtParam including solubility and disulphide bonds in the SCRATCH server. The vaccine 3D structure was built using I-TASSER and refined in ModRefine. Docking between JUNV glycoprotein (PDB ID:5NUZ) with a built vaccine revealed a balanced docked complex visualized in the Drug Discovery studio, identified 280 hydrogen bonds between them. The docking score of − 15.5 kcal/mol was determined in the MM/GBSA analysis in HawkDock. MD simulations employed using the GROMACS at 20 ns resulted in minimal deviation and fewer fluctuations, particularly with high hydrogen bond-forming residues.
Conclusion
However, these findings present a potential vaccine for developing against JUNV glycoprotein after validating the epitopes and 3D vaccine construct through in silico methods. Therefore, further investigation in the wet laboratory is necessary to confirm the potentiality of the predicted vaccine.
Similar content being viewed by others


Background
The Junín virus (JUNV) is a member of the Arenaviridae family, renowned for causing Argentine haemorrhagic fever (AHF), which is a severe endemic infection prevalent in populations residing around agricultural areas in Argentina [1]. The first cases were reported in the 1950s during the unleashed propagation of Calomys musculus. Since then, hundreds of cases have been registered annually. Humans will become victims by the inhalation of rodent aerosols or excreta, generally in the harvest season, so it is a rodent-borne virus [2]. Common symptoms of AHF include flu-like signs, such as headache, malaise, and fever. Primary infection sites are the lungs and then circulate to parenchymal tissues [3]. The final stage of AHF shows neurologic and haemorrhagic complications. The World Health Organization (WHO) has classified AHF as an emerging disease, warranting immediate research to design antiviral agents and vaccine targeting the virus components.
JUNV’s genome consists of two single-stranded RNA segments: a small (S) segment of 3.4 kb and a large (L) segment of 7.2 kb. The S segment translates in the cytoplasm giving rise to nucleoproteins and glycoprotein precursors that mature into a glycoprotein complex after cleavage by cellular proteases. The glycoprotein is crucial for viral attachment to host cells, initiating the viral components' entry into the host cell [4,5,6]. Glycoprotein formation occurs in the endoplasmic reticulum and then infects parenchymal cells through circulation [4]. Glycoprotein also plays a role in cell pathogenicity [7].
In the late 1990s, an effective live-attenuated vaccine was developed, significantly reducing AHF incidence. However, there are no effective remedies to counter viral infection among victims [8]. Later, the application of immune plasma therapy to neutralize the antigens decreased the 1% fatality rate if treated before eight days of the onset of symptoms [9,10,11,12]. The FDA-approved nucleoside analogue ribavirin inhibits JUNV polymerase and serves as an antiviral, but its use is complicated due to side effects and less efficacy [12, 13]. Numerous small molecule (drugs) antiviral compounds have been reported as antagonists to JUNV in both in vitro [14,15,16,17] and in animal models [18, 19]. In silico approaches, such as molecular docking, have been employed to identify the potent drugs from the FDA against the glycoprotein of the Junin virus, revealing MK-3207 and dihydro-ergotamine [17]. Additionally, the live-attenuated vaccine strain Candid #1 (Can) has shown promise in inhibiting glycoprotein spread and reducing infections [18].
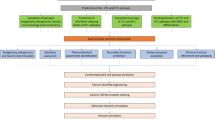
Until now, the experiments have primarily focussed on neutralized live-attenuated vaccines, but in this study, we aim to design a new vaccine specifically targeting the glycoprotein. Firstly, we have chosen the mature glycoprotein sequence in identifying and designing a new vaccine through immunoinformatics approaches such as predicting the B cell and T cell epitopes. Next, validating the peptide sequences to ensure the accuracy and reliability of epitopes. Subsequently, designing the vaccine by incorporating suitable adjuvants and linkers and analysing the physicochemical properties. Furthermore, detailed evaluations of the secondary and tertiary structural features of the designed vaccine sequence followed by refining the 3D structure. Furthermore, we checked the binding affinity score and molecular dynamic simulation of the designed vaccine. We presented the detailed methodology followed during this study in a pictorial format, making it visually accessible and facilitating a better understanding in Fig. 1.

The methodology followed in this study
Methods
Sequence retrieval
The sequence of Junín virus glycoprotein precursor was retrieved from the National Centre for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov/nuccore/MZ408914.1) [19] of 485 length peptide sequence. NCBI provides various tools and a repository database to access the sequence deposited in it.
Prediction of B cell and T cell [MHC I (TC/CD4+), MHC II (TH/CD8+)] epitopes
To predict B cell linear epitopes, we used the Immune Epitope Database (IEDB) (http://tools.immuneepitope.org/bcell/) and employed Bepipred Linear Epitope prediction 2.0 with a threshold value of 0.50 [20]. This prediction method is based on the input fasta format of the query protein sequence, and the server utilizes the random forest algorithm to distinguish between epitopes and non-epitope sequences found in crystal structures.
For predicting cytotoxic T cells/Class 1/CD4+ epitopes, we used artificial neural network 4.0 [21] and specified human MHC alleles as the source or reference to get the IC50 values of each predicted sequence in the IEDB Analysis Resource (http://tools.iedb.org/mhci/), which determines the subsequence’s binding ability with the specific MHC class I. Helper T cells/Class II/CD8+ epitopes predictions were carried out with the NN-align 2.3 (NetMHCII) method [22, 23] with the human HLA-DR data set and all MHC II allele sequences between 12 and 18 mers in length. The resulting output provided IC50 values for predicted epitopes in the IEDB Analysis Resource (http://tools.iedb.org/mhcii/).
Validation of epitopes
The predicted epitopes were subjected to validation for allergenicity, antigenicity, and toxicity. Allergenicity refers to the ability of an antigen to induce abnormal and hyperresponsive [23]. AllerTOP v. 2.0 (http://www.ddg-pharmfac.net/AllerTOP/) was utilized for allergenicity prediction, employing the k-nearest neighbour algorithm on a training set of 2427 allergens and 2427 allergens, based on ACC uniform length and QSAR with different lengths [24]. Antigenicity, on the other hand, triggers the immune response and describes the ability to bind paratopes [25]. VaxiJen 2.0 (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) server used to predict the antigens and non-antigens [26]. This server was developed based on independent alignment methods with auto cross-covariance (ACC) of amino acid sequences transformation into principal vector properties to predict antigens and non-antigens. To access the peptides' toxicity, the ToxinPred server (https://webs.iiitd.edu.in/raghava/toxinpred/multi_submit.php) [27] was used. It utilizes the support vector machine (SVM) method with a threshold of 0.5 and an E value of 0.01 to predict the toxic and non-toxic peptides. Furthermore, population coverage analysis (http://tools.iedb.org/population/) [27] was performed to select the world population for MHC class I and MHC class II, which covers 3245 alleles from 16 geographical areas, 21 various ethnicities, and 115 countries.
Designing and characterization of structural vaccine
A serial arrangement of the vaccine was designed with beta-defensin 114 as an adjuvant and EAAK, AAY, GPGPG, and KK as flexible linkers [28, 29]. Epitope inclusion linkers and adjuvants were aimed to enhance protein stability and immunogenicity. To validate the designed vaccine, various physicochemical properties were calculated using ProtParam (https://web.expasy.org/protparam/) [30]. Antigenicity was determined using VaxiJen v 2.0 [26], while solubility with SolPro [31]. The presence of disulphide bonds was analysed through DLpro [32] through the SCRATCH web server [33] (https://scratch.proteomics.ics.uci.edu/). Allergenicity prediction was performed through AllerTOP v. 2.0 [24].
Secondary and tertiary structure assessment of antibody
Psipred [34] (http://bioinf.cs.ucl.ac.uk/psipred/) was designed based on the two feed-forward neural networks using the output from Position-Specific Iterated (PSI) – Blast. The SOPMA Server [35] (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_sopma.html) uses amino acid sequences by the self-optimized prediction method to predict the secondary structure features of the protein.
For in silico studies requiring tertiary structure of the protein 3D protein models using were generated using the servers such as Iterative Threading ASSEmbly Refinement (I-TASSER) [36] (https://zhanggroup.org/I-TASSER/). The prediction is based on the multiple threading approaches from the protein data bank through Local Meta-Threading Server v 3.0. The complete model was built by iterative template-based fragment assembly. The generated models were refined using the ModRefiner [37] (https://zhanggroup.org/ModRefiner/) servers until they achieved Ramachandran-favoured regions greater than 90%.
Molecular docking and MM/GBSA evaluation
The glycoprotein (GP) complex with the Fab antibody of the Junin virus was obtained from the Protein Data Bank (PDB ID: 5NUZ) (https://www.rcsb.org/) and determined using the X-ray diffraction method. The raw protein structure was made by eliminating the extra bound ligands, water, and het atoms followed by saving them in.pdb format for docking. The GP and the designed antibody were docked in the ClusPro protein–protein docking server (https://cluspro.bu.edu/home.php) [38], which was different from the protein–ligand docking that we performed in our previous studies [39].
Subsequently, the docked complex underwent Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) analysis to determine various interaction energies such as binding affinity, Vander Waal, electrostatic, polar solvation-free energy, and solvation-free energy based on the empirical methodologies after minimizing the complex in the ff02 force field over the HawkDock server (http://cadd.zju.edu.cn/hawkdock/) [40].
Molecular dynamic (MD) simulations
The MD simulations were performed on the refined tertiary structure of the antibody in a water medium using GROMACS [41] (https://simlab.uams.edu/index.php) from the WebGRO sim lab UAMS. GROMOS96 43a1 force field [42] initially used the TIP4P solvation model, to build the triclinic box and maintained 0.15 molarity of NaCl at pH 7.4. Using the steepest descent type of energy minimization 800 kJ/mol at 10,000 steps with NVT and NPT equilibration at temperature 300 k and pressure at 1 atmosphere. The final MD was performed by a leap-frog integer method at 20 ns of time.
Results
Sequence retrieval
The NCBI Virus accession number for the glycoprotein sequences of the GenBank ID: MZ408914.1, represents the glycoprotein sequence of the S segment of 485 length peptide sequence linear RNA genome isolated from the Argentinian mammarenavirus on Feb 2022.
Prediction and validation of B cell and T cell epitopes
The B cell and T cell epitopes were predicted using different algorithms from the IEDB, and their respective antigenicity (A > 0.5), allergenicity (+), and toxicity (N) were manually analysed and curated (eliminated the non-antigenic (NA), allergenic (−), and toxic (T) peptides). Initially, fourteen B cell epitopes were retrieved as outputs, and they were curated based on properties such as antigenicity, allergenicity, and toxicity. Finally, four peptide sequences were selected for the vaccine construction in Table 1, after removing the duplicates. Based on the threshold values, numerous MHC class I and class II epitopes were obtained. Furthermore, filtered based on IC50 values (≤ 20), were selected followed by antigenic, non-allergen, and non-toxic peptides used in designing the antibody sequence in Table 2. Population coverage analysis for MHC I and MHC II epitopes was employed to estimate the potential target population for the predicted epitopes used in designing the vaccine, resulting in the best coverage value of 97.28% world population (Additional file 2: Table S1 and Figure S1).
Designing and characterization of antibody structure
Before designing the vaccine, a suitable adjuvant, beta-defensin was selected, which is widely used. Additionally, appropriate linkers were exploited to connect intra B cells with KK, MHC I with AAY, and MHC II epitopes with GPGPG linkers and among them during vaccine designing. An EAAAK rigid linker, capable of forming an alpha helix at the amine terminal used to link the B cell epitopes and the adjuvant. The GPGPG linker was used between the MHC I epitopes and B cell epitopes. An AAY linker was employed to join the MHC I and MHC II epitopes followed by six histidine tags (6H) at the end in Fig. 2.

The sequential arrangement of epitopes in designing the vaccine
The designed antibody underwent physicochemical analysis of the nine descriptor values determined through ExPASY ProtParam. The vaccine comprises 431 residues with 47,835.77 Daltons molecular weight (M.wt.). The isoelectric constant point (pI) of the designed antibody was found to be 9.65. The negatively (Asp + Glu) [−R] and positively (Arg + Lys) [+ R] charged residues were 29 and 57, respectively. The extinction coefficient (EC) value at 280 nm was 36,258 M−1 cm−1. The instability index (II) was 34.87, a value greater than 40 refers to unstable, and a value less than 40 stability. The aliphatic index (AI) was 81.25 and the grand average of hydropathicity (GRAVY) of − 0.254 in Table 3.
The descending order of the amino acid types in the antibody was: Polar > non-polar > basic > aromatic > acidic in Fig. 3. The constructed vaccine was validated as antigenic, non-allergenic, non-toxic, and had a solubility of 0.92. Moreover, it could form six disulphide bonds between the cysteine residues in Table 4 and Additional file 2: Table S2.

Different amino acid type content in the designed vaccine sequence
Secondary and tertiary structure assessment of antibody
The designed antibody fasta sequence was submitted to the SOPMA server for secondary structure prediction. The results indicated that the antibody comprises 26.68% of alpha helices, 41.07% of random coils, 25.99% of extended strands, and 6.26% of beta turns in Table 5. Figure 4, obtained from PSIPRED, exhibits the secondary structure confidence score and provides additional pictorial information about the secondary structure, as depicted in Additional file 2: Figure S2.

The confidence level of prediction of the secondary structure via PSIPRED
The tertiary model obtained from the I-TASSER, though constructed upon validating in the Ramachandran plot, does not have enough amino acids in the allowed regions, leading to a low confidence score (C-score) of − 2.17. The C-Score ranges from − 5 to 2, where a higher value indicates a better model. Based on the confidence score, template modelling (TM-score) and root-mean-square deviation (RMSD) values were calculated to assess the structural similarity between the model and the actual protein. That resulted in a TM-Score of 0.46 ± 0.15 and 12.2 ± 4.4 [43].
The RMSD quantifies the similarity between two superimposed atomic coordinates [44]. The model was sent for structure refinement in the Galaxy refiner, and the results are shown in Additional file 2: Table S3. Based on the Global distance test (GDT) score, RMSD value, molprobity, clash score, rotamers value, and Ramachandran-favoured regions, model 1 was selected as a vaccine which was furthermore analysed. The GDT-HA represents the high-resolution value of the structure, and the MolProbity value is a log-weighted value determined from scores such as clash score, Ramachandran-favoured percentage, and bad side-chain rotamers [45]. Ramachandran’s favoured percentage regions have residues of 90.9% in the most favoured regions of the built 3D antibody. Overall, the selected model was refined and met the criteria for a suitable vaccine candidate, based on the various evaluation structural parameters and properties.
Molecular docking and MM/GBSA evaluation
The interactions between Junín glycoprotein and the designed antibody were analysed through the ClusPro server, and a balanced complex is retrieved in Fig. 5. The interactions between the residues were visualized in the drug discovery studio. Before this, the vaccine formed a total of nine hundred fourteen interactions with the glycoprotein. Including four S–S bonds, twenty-five electrostatic bonds, one-hundred eighty-three hydrophobic bonds, and seven hundred-two hydrogen bonds. Hydrogen bond-forming interactions play a prominent role in drug availability and enhancing the binding chances. Furthermore, the distance criteria of less than two angstroms (< 2 Å) interaction distance among the hydrogen bonds were 280 (Additional file 1).

The docked complex of JUNV glycoprotein (Chain C and L) with the designed vaccine (Chain A)
Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) was employed in determining the free energy between the glycoprotein and the designed vaccine (protein–protein interaction). Extensive methodologies in calculating the interaction energies include receptor’s grid identification, pre-processing the input files, T leap function, scoring various energies, and scoring. Briefly van der Waals (VDW) energy was − 151.33 kcal/mol, electrostatic energy (ELE) was − 272.63 kcal/mol, polar solvation-free energy (GB) was 424.39 kcal/mol, and solvation-free (SA) energy was − 15.93 kcal/mol. The total binding free energy of the complex was − 15.5 kcal/mol represented in Table 6. Overall, the analysis indicates strong and favourable interactions between the Junin glycoprotein and the designed vaccine, with a significant binding free energy, suggesting the potent vaccine’s effectiveness in combating the virus.
Molecular dynamic simulations
Understanding the vaccine’s conformational relative stability of the backbone atoms using the RMSD measure provides valuable insights since it measures the deviations of the extent atoms from the starting point. Less deviation (low RSMD) value signifies a better stable protein structure. For the constructed vaccine, the average lowest RMSD value was 0.24 nm in Fig. 6A. However, there was a slight increase in atomic deviations to 0.6 nm at 0.3 ns. This increase in RMSD values between 0.6 to 0.8 nm from 0.3 to 15 ns can be attributed to the modelled vaccine structural interactions optimization.

The molecular dynamic simulations of the 3D vaccine in water. A Root-mean-square deviation (RMSD) computes the average distance between the backbone atoms of starting structure (reference structure) with simulated structures (frame by frame) when superimposed. B Root-mean-square fluctuation (RMSF) computes fluctuations (standard deviation) of atomic positions of each amino acid (residues) in the trajectory [X-axis = Time; Y-axis = Residue number]. C Hydrogen interactions forming residues
Furthermore, the protein stability evaluated by root-mean-square fluctuations (RMSFs), which measures the protein residue’s elasticity and binding site adaptation phenomena, was determined. The vaccine, RMSF contains evident residue cluster fluctuations. On average, there were two fluctuations for every hundred residues. Residues 200–320 showed high fluctuations with significant up and down levels. Maximum fluctuations were observed in the 390–400 residues range, while minimum fluctuations between 280 and 389 in Fig. 6B.
Hydrogen bond trajectories retained during the MD simulation were studied to evaluate the strength and binding affinity of the complex. The designed vaccine was found to form several hydrogen bonds with an average of 140 hydrogen bonds when interacting with the antigenic protein. The ability to form multiple hydrogen bonds was crucial in inhibiting the target protein, thereby strengthening the complex’s free energy in Fig. 6C. Overall, analysis of RMSD, RMSF, and hydrogen bond trajectories provides valuable information about the stability and interactions of the designed vaccine, which were essential factors in determining its potential effectiveness as a vaccine.
Discussion
The general effective possibilities that are being examined are to control the dispense of Argentine haemorrhagic fever (AHF) by the design and development of suitable vaccines. The duration of the vaccine development from experimental studies to clinical trial studies takes an extensive period. In this modern era, due to the advancement of system applications in biology, numerous epitopes predicting epitopes are made available using various machine learning algorithms. Among them, multi-epitope-based vaccine designs employing predicting B cell and T cell epitopes have made a remarkable trend in bioinformatics. In addition, proper attention is required to develop a safe and viable vaccine.
To date, the primary approach to prevent the infection rate is neutralized monoclonal antibodies (MABs). Research on the MABs extracted from animal models (mouse) shows that the glycoprotein JUNV is involved in mimicking the human transferrin receptor 1 (hTfR1) during the binding process. This glycoprotein acts as a primary target and plays a predominant role in the prevention and treatment of infection [46]. The vaccine design according to the sequence-specific will act efficiently in inhibiting the antigen and will also be helpful in further mutational studies considering the evolution of the virus variants.
Three epitope types (B cell, TC, and TH) predictions were performed using multiple servers including experimentally based HLA class I and HLA class II alleles [47]. All predicted epitopes were validated based on antigenicity/non-antigenicity, allergenicity/non-allergenicity, and toxicity/non-toxicity and shared maximum population coverage across the world. The number of B cell epitopes obtained after screening was less, indicating fewer interactions. Considering the T cell epitope number obtained, the cell-mediated immunity and immune response generated were high and long-lasting. A commonly used adjuvant (beta-defensin) was selected in designing and the linkers were added appropriately in Fig. 2. To ensure safety and efficacy, the designed vaccine over-checked their non-allergenicity, antigenicity, and non-toxicity. Furthermore, physicochemical, solubility and disulphide bond parameters were checked to determine the stability of the vaccine in Table 4 and Additional file 2: Table S2. This comprehensive validation paves potential applications in immunization against the target antigen.
The physicochemical properties in Table 3 and Fig. 3 include the pI as the pH at which no electrical charge is present on the molecule or the total number of negative and positive charges are equal [30]. The isoelectric focusing technique is performed based on the pI values for separating the molecule from the complex [48]. These values help to isolate the respective protein of interest in the wet lab experiments upon digestion. The glycoprotein’s pI of 9.65 indicates that for wet-lab studies the experimental setup is to be maintained a basic environment during extraction. EC is defined as the amount of light absorbed per mole of protein at a specific wavelength of light. The protein’s EC value is calculated from the composition of tryptophan, tyrosine, and cysteine residues because these amino acids contribute significantly to measuring the protein’s optical density in the 276–282 nm range [30]. Protein–protein and protein-ligands quantitative study can be understood through the EC values [49]. The EC value exhibited was high representing a good sequence for further studies. II indicates the protein stability under both in vivo and in vitro conditions. Proteins with II greater than 40 are considered unstable proteins, while II less than 40 are stable [50]. II of the designed vaccine was 34.87 inferring the stability. AI is another parameter that describes protein stability at temperatures. AI is defined as the relative volume occupied by aliphatic side chains like alanine (Ala), valine (Val), leucine (Leu), and isoleucine (Ile) [30, 48]. The high value of AI indicates the increase in the thermostability nature of the protein which is an additive factor for wet lab studies. The Aliphatic index (AI) of the vaccine sequence is 81.25 indicating the thermostability character. GRAVY value ranges from − 4 to + 4 indicating the hydrophilic and hydrophobic nature of the proteins [51]. The low GRAVY range indicates the possibility of being a globular (hydrophobic) protein rather than membranous (hydrophilic). Thus, the vaccine contains a globular protein residue with a value of − 0.254.
The tertiary structure of the vaccine has been generated from the I-TASSER and further processed in ModRefiner to achieve optimum descriptor values such as GDT-HA, RMSD, Molprobity, C-score, poor rotamers, and Ramachandran-favoured regions greater than or equal to 90% in Additional file 2: Table S3, aids to validate the designed structure all of which are crucial in validating the structural integrity [52, 53]. Subsequently, molecular docking of JUNV glycoprotein and the construct was performed to achieve a balanced complex model from the ClusPro server in Fig. 5. The post-docking analysis, specifically the MM/GBSA method in Table 6, of the docked complex resulted in favourable interaction energies within the docked complex. Moreover, MD simulations on the vaccine in water in Fig. 6 have indicated that the designed construct exhibited notable stability without higher deviations and fluctuations with maximum hydrogen bond forming ability that emphasizes the vaccine stability. These findings collectively underscore the designed vaccine’s reliability and robustness for potential application in combating the JUNV.
Conclusion
In this study, we focussed on vaccine design based on the glycoprotein of the JUNV on employed immunoinformatics approaches to identify antigenic peptides because of the novelty of identifying the peptides using machine learning tools. The vaccine was constructed after selecting the antigenic, nonallergic, and non-toxic and deleting the duplicate and overlapping sequences. The vaccine was designed using 4 B cell, 9 MHC class I, and 8 MHC class II T cell epitopes with appropriate linkers and adjuvant. The tertiary model was generated and refined to meet suitable specific criteria especially Ramachandran-favoured regions greater than 90%. After the physicochemical properties’ determination, molecular docking of JUNV glycoprotein with the designed vaccine performed a balanced complex model chosen for analysing the interactions among them showed 280 hydrogen bond forming residues with radii less than 2 angstroms. MM/GBSA methodology exhibited − 15.5 kcal/mol of binding free energy of the complex. The MD simulations of the vaccine that were performed exhibited fewer values of fluctuations and deviations that signify the vaccine's stability. However, the present work was based on in silico methodologies with promising results that will be evident in the wet lab works to justify the findings.
Availability of data and materials
The data that support the findings of this study are available from the corresponding author, upon reasonable request.
References
Maiztegui JI (1975) Clinical and epidemiological patterns of Argentine haemorrhagic fever. Bull World Health Organ 52:567–575
Maiztegui JI, McKee KT Jr, Barrera Oro JG, Harrison LH, Gibbs PH, Feuillade MR, Enria DA, Briggiler AM, Levis SC, Ambrosio AM, Halsey NA, Peters CJ (1998) Protective efficacy of a live attenuated vaccine against Argentine hemorrhagic fever AHF Study Group. J Infect Dis 177(2):277–283. https://doi.org/10.1086/514211
Gómez RM, Jaquenod de Giusti C, Sanchez Vallduvi MM, Frik J, Ferrer MF, Schattner M (2011) Junín virus. A XXI century update. Microbes Infect 13:303–311. https://doi.org/10.1016/j.micinf.2010.12.006
Nunberg JH, York J (2012) The curious case of arenavirus entry, and its inhibition. Viruses 4:83–101. https://doi.org/10.3390/v4010083
Burri DJ, Palma JR, Kunz S, Pasquato A (2012) Envelope glycoprotein of arenaviruses. Viruses 4:2162–2181. https://doi.org/10.3390/v4102162
York J, Nunberg JH (2006) Role of the stable signal peptide of Junín arenavirus envelope glycoprotein in pH-dependent membrane fusion. J Virol 80(15):7775–7780. https://doi.org/10.1128/JVI.00642-06
Koma T, Huang C, Coscia A, Hallam S, Manning JT, Maruyama J, Walker AG, Miller M, Smith JN, Patterson M, Abraham J, Paessler S (2021) Glycoprotein N-linked glycans play a critical role in arenavirus pathogenicity. PLoS Pathog 17(3):e1009356. https://doi.org/10.1371/journal.ppat.1009356
Maiztegui JI, McKee KT Jr, Barrera Oro JG, Harrison LH, Gibbs PH, Feuillade MR, Enria DA, Briggiler AM, Levis SC, Ambrosio AM, Halsey NA, Peters CJ (1998) Protective efficacy of a live attenuated vaccine against Argentine hemorrhagic fever AHF Study Group. J Infect Dis 177(2):277–283. https://doi.org/10.1086/514211
Maiztegui JI, Fernandez NJ, de Damilano AJ (1979) Efficacy of immune plasma in treatment of Argentine haemorrhagic fever and association between treatment and a late neurological syndrome. Lancet (London, England) 2(8154):1216–1217. https://doi.org/10.1016/s0140-6736(79)92335-3
Sepúlveda CS, García CC, Damonte EB (2018) Antiviral activity of A771726, the active metabolite of leflunomide, against Junín virus. J Med Virol 90(5):819–827. https://doi.org/10.1002/jmv.25024
Enria DA, Maiztegui JI (1994) Antiviral treatment of Argentine hemorrhagic fever. Antiviral Res 23(1):23–31. https://doi.org/10.1016/0166-3542(94)90030-2
Enria DA, Briggiler AM, Sánchez Z (2008) Treatment of Argentine hemorrhagic fever. Antiviral Res 78(1):132–139
Charrel RN, Coutard B, Baronti C, Canard B, Nougairede A, Frangeul A, Morin B, Jamal S, Schmidt CL, Hilgenfeld R, Klempa B, de Lamballerie X (2011) Arenaviruses and hantaviruses: from epidemiology and genomics to antivirals. Antiviral Res 90(2):102–114
Rathbun JY, Droniou ME, Damoiseaux R, Haworth KG, Henley JE, Exline CM, Choe H, Cannon PM (2015) Novel arenavirus entry inhibitors discovered by using a minigenome rescue system for high-throughput drug screening. J Virol 89(16):8428–8443. https://doi.org/10.1128/JVI.00997-15
Chou YY, Cuevas C, Carocci M, Stubbs SH, Ma M, Cureton DK, Chao L, Evesson F, He K, Yang PL, Whelan SP, Ross SR, Kirchhausen T, Gaudin R (2016) Identification and characterization of a novel broad-spectrum virus entry inhibitor. J Virol 90(9):4494–4510. https://doi.org/10.1128/JVI.00103-16
Gowen BB, Juelich TL, Sefing EJ, Brasel T, Smith JK, Zhang L, Tigabu B, Hill TE, Yun T, Pietzsch C, Furuta Y, Freiberg AN (2013) Favipiravir (T-705) inhibits Junín virus infection and reduces mortality in a guinea pig model of Argentine hemorrhagic fever. PLoS Negl Trop Dis 7(12):e2614. https://doi.org/10.1371/journal.pntd.0002614
Malhotra H, Kumar A, Afaq Y (2022) Molecular docking analysis of FDA approved drugs with the glycoprotein from Junin and Machupo viruses. Bioinformation 18(2):119–126. https://doi.org/10.6026/97320630018119
Seregin AV, Yun NE, Miller M, Aronson J, Smith JK, Huang C, Manning JT, de la Torre JC, Paessler S (2015) The glycoprotein precursor gene of Junin virus determines the virulence of the Romero strain and the attenuation of the Candid #1 strain in a representative animal model of Argentine hemorrhagic fever. J Virol 89(11):5949–5956. https://doi.org/10.1128/JVI.00104-15
Roman-Sosa G, Leske A, Ficht X, Dau TH, Holzerland J, Hoenen T, Beer M, Kammerer R, Schirmbeck R, Rey FA, Cordo SM, Groseth A (2022) Immunization with GP1 but not core-like particles displaying isolated receptor-binding epitopes elicits virus-neutralizing antibodies against junín virus. Vaccines 10(2):173. https://doi.org/10.3390/vaccines10020173
Jespersen MC, Peters B, Nielsen M, Marcatili P (2017) BepiPred-20: improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res 45(1):24–29. https://doi.org/10.1093/nar/gkx346
Andreatta M, Nielsen M (2016) Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics (Oxford, England) 32(4):511–517. https://doi.org/10.1093/bioinformatics/btv639
Jensen KK, Andreatta M, Marcatili P, Buus S, Greenbaum JA, Yan Z, Sette A, Peters B, Nielsen M (2018) Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology 154(3):394–406
Zhang J, Tao A (2015) Antigenicity, immunogenicity, allergenicity. Allergy Bioinform 8:175–186. https://doi.org/10.1007/978-94-017-7444-4_11
Dimitrov I, Bangov I, Flower DR, Doytchinova I (2014) AllerTOP vol 2–a server for in silico prediction of allergens. J Mol Model 20(6):2278. https://doi.org/10.1007/s00894-014-2278-5
Van Regenmortel MHV (1992) Protein antigenicity. Mol Biol Rep 16(3):133–138. https://doi.org/10.1007/BF00464700
Doytchinova IA, Flower DR (2007) VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics 8:4. https://doi.org/10.1186/1471-2105-8-4
Bui HH, Sidney J, Dinh K, Southwood S, Newman MJ, Sette A (2006) Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinform 7:153. https://doi.org/10.1186/1471-2105-7-153
Chen X, Zaro JL, Shen WC (2013) Fusion protein linkers: property, design and functionality. Adv Drug Deliv Rev 65(10):1357–1369. https://doi.org/10.1016/j.addr.2012.09.039
Li G, Huang Z, Zhang C, Dong BJ, Guo RH, Yue HW, Yan LT, Xing XH (2016) Construction of a linker library with widely controllable flexibility for fusion protein design. Appl Microbiol Biotechnol 100(1):215–225. https://doi.org/10.1007/s00253-015-6985-3
Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, Hochstrasser DF (1999) Protein identification and analysis tools in the ExPASy server. Meth Mol Biol (Clifton, N.J.) 112:531–552. https://doi.org/10.1385/1-59259-584-7:531
Magnan CN, Randall A, Baldi P (2009) SOLpro: accurate sequence-based prediction of protein solubility. Bioinformatics (Oxford, England) 25(17):2200–2207. https://doi.org/10.1093/bioinformatics/btp386
Cheng J, Saigo H, Baldi P (2006) Large-scale prediction of disulphide bridges using kernel methods, two-dimensional recursive neural networks, and weighted graph matching. Proteins 62(3):617–629. https://doi.org/10.1002/prot.20787
Cheng J, Randall AZ, Sweredoski MJ, Baldi P (2005) SCRATCH: a protein structure and structural feature prediction server. Nucleic Acids Res 33:W72–W76. https://doi.org/10.1093/nar/gki396
McGuffin LJ, Bryson K, Jones DT (2000) The PSIPRED protein structure prediction server. Bioinformatics (Oxford, England) 16(4):404–405. https://doi.org/10.1093/bioinformatics/16.4.404
Geourjon C, Deléage G (1995) SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput Appl Biosci: CABIOS 11(6):681–684. https://doi.org/10.1093/bioinformatics/11.6.681
Zheng W, Zhang C, Li Y, Pearce R, Bell EW, Zhang Y (2021) Folding non-homologous proteins by coupling deep-learning contact maps with I-TASSER assembly simulations. Cell Reports Meth 1(3):100014. https://doi.org/10.1016/j.crmeth.2021.100014
Xu D, Zhang Y (2011) Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys J 101(10):2525–2534. https://doi.org/10.1016/j.bpj.2011.10.024
Comeau SR, Gatchell DW, Vajda S, Camacho CJ (2004) ClusPro: a fully automated algorithm for protein-protein docking. Nucleic Acids Res 32:W96–W99. https://doi.org/10.1093/nar/gkh354
Praveen M, Morales-Bayuelo A (2023) Drug designing against VP4, VP7 and NSP4 of rotavirus proteins – insilico studies. Moroccan J Chem 11:729–741. https://doi.org/10.48317/IMIST.PRSM/MORJCHEM-V11I3.40088
Weng G, Wang E, Wang Z, Liu H, Zhu F, Li D, Hou T (2019) HawkDock: a web server to predict and analyze the protein-protein complex based on computational docking and MM/GBSA. Nucleic Acids Res 47(W1):W322–W330. https://doi.org/10.1093/nar/gkz397
Abraham MJ, Murtola T, Schulz R, Pali S, Smith JC, Hess B, Lindahi E (2015) GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2:19–25. https://doi.org/10.1016/j.softx.2015.06.001
Pol-Fachin L, Fernandes CL, Verli H (2009) GROMOS96 43a1 performance on the characterization of glycoprotein conformational ensembles through molecular dynamics simulations. Carbohyd Res 344(4):491–500. https://doi.org/10.1016/j.carres.2008.12.025
Zhang Y, Skolnick J (2004) Scoring function for automated assessment of protein structure template quality. Proteins 57(4):702–710. https://doi.org/10.1002/prot.20264
Kufareva I, Abagyan R (2012) Methods of protein structure comparison. Meth Mole Biol (Clifton, N.J.) 857:231–257. https://doi.org/10.1007/978-1-61779-588-6_10
Williams CJ, Headd JJ, Moriarty NW, Prisant MG, Videau LL, Deis LN, Verma V, Keedy DA, Hintze BJ, Chen VB, Jain S, Lewis SM, Arendall WB 3rd, Snoeyink J, Adams P, Lovell SC, Richardson JS, Richardson DC (2018) MolProbity: more and better reference data for improved all-atom structure validation. Prot Sci: A Publ Prot Soci 27(1):293–315. https://doi.org/10.1002/pro.3330
Ng WM, Sahin M, Krumm SA, Seow J, Zeltina A, Harlos K, Paesen GC, Pinschewer DD, Doores KJ, Bowden TA (2022) Contrasting modes of new world arenavirus neutralization by immunization-elicited monoclonal antibodies. mBio 13(2):e0265021. https://doi.org/10.1128/mbio.02650-21
Greenbaum J, Sidney J, Chung J, Brander C, Peters B, Sette A (2011) Functional classification of class II human leukocyte antigen (HLA) molecules reveals seven different supertypes and a surprising degree of repertoire sharing across supertypes. Immunogenetics 63(6):325–335. https://doi.org/10.1007/s00251-011-0513-0
Pergande MR, Cologna SM (2017) Isoelectric point separations of peptides and proteins. Proteomes 5(1):4. https://doi.org/10.3390/proteomes5010004
Mohan R, Venugopal S (2012) Computational structural and functional analysis of hypothetical proteins of Staphylococcus aureus. Bioinformation 8(15):722–728. https://doi.org/10.6026/97320630008722
Guruprasad K, Reddy BV, Pandit MW (1990) Correlation between stability of a protein and its dipeptide composition: a novel approach for predicting in vivo stability of a protein from its primary sequence. Protein Eng 4(2):155–161. https://doi.org/10.1093/protein/4.2.155
Kyte J, Doolittle RF (1982) A simple method for displaying the hydropathic character of a protein. J Mol Biol 157(1):105–132. https://doi.org/10.1016/0022-2836(82)90515-0
Praveen M, Ullah I, Buendia R, Khan IA, Sayed MG, Kabir R, Bhat MA, Yaseen M (2024) Exploring Potentilla nepalensis Phytoconstituents: integrated strategies of network pharmacology, molecular docking, dynamic simulations, and MMGBSA analysis for cancer therapeutic targets discovery. Pharmaceuticals 17:134. https://doi.org/10.3390/ph17010134
Praveen M (2024) Characterizing the West Nile Virus’s polyprotein from nucleotide sequence to protein structure - Computational tools. J Taibah Univer Med Sci 19(2):338–350. https://doi.org/10.1016/j.jtumed.2024.01.001
Acknowledgements
Not applicable.
Funding
No funding is provided by any government agency or non-government organization.
Author information
Authors and Affiliations
Contributions
Not applicable.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1. Table S1.
Population coverage analysis of combined MHC I and MHC II. Table S2. Predicted disulphide bonds (cysteine pairs) ordered by probability in descending order. Table S3. Vaccine tertiary models generated by ModRefiner. Figure S1. The population coverage epitopes pictorial presentation. Figure S2. The properties of vaccine secondary structure.
Additional file 2.
Interacting residues forming hydrogen bonds between the JNV glycoprotein and designed vaccine.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Praveen, M. Multi-epitope-based vaccine designing against Junín virus glycoprotein: immunoinformatics approach. Futur J Pharm Sci 10, 29 (2024). https://doi.org/10.1186/s43094-024-00602-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43094-024-00602-8