
Abstract
Background
A simple, rapid, sensitive and selective stability-indicating (RP-HPLC) method is suggested for the determination of Mupirocin calcium in bulk drug and in pharmaceutical formulation. Mupirocin calcium was eluted from a PrincetoneSPHER-100 C8 (250 × 4.6 mm, 5 µm) column with mobile phase consisting of methanol and water (75:25 v/v) pH adjusted to 4 with acetic acid. The gradient was optimized with a flow rate of 1 mL/min and a wavelength of 221 nm.
Result
The complete analytical method validation was successfully carried out as per ICH guidelines. The retrieval study was carried out at 80% to 120% level of working concentration, and results were in the range of 99 to 101%. The linearity was proven in range of 4–24 µg/mL of working concentration with linear regression curve (R2 = 0.999) with limits of detection (LOD) and quantitation (LOQ) being 0.35 and 1.08 µg/mL, respectively. The retention time for Mupirocin calcium was 5.09 min. The method shows good recoveries and intra-day and inter-day relative standard deviations were less than 2%. Validation parameters as ruggedness and robustness were also determined as per ICH guidelines and were found to be satisfactory. For stability study, the drug was exposed to various stress conditions such as acid, base, oxidation and sunlight as per recommendations of ICH guidelines.
Conclusion
The developed HPLC method could be successfully used for the estimation of Mupirocin calcium in bulk and in Pharmaceutical formulation. The high recovery and low relative standard deviation confirm the suitability of proposed method that can be employed for the routine analysis in bulk and Pharmaceutical formulation.
Similar content being viewed by others


Background
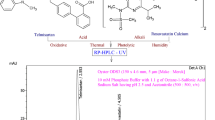
Mupirocin calcium [MUP] (Fig. 1) is chemically (αE, 2S, 3R, 4R, 5S) -5- [(2S, 3S, 4R, 5S) -5-[(2S, 3S, 4S, 5S)- 2,3-Epoxy-5-methylhexyl] tetra hydro 3,4-dihydro-β-methyl-2H-pyran-2-crotonic acid, ester with 9-hydroxynonanoic acid, calcium salt (2:1), dehydrate [1]. The drug acts by inhibiting bacterial isoleucyl-tRNA synthetase [2, 3] thereby blocking protein synthesis and indirectly inhibiting RNA synthesis. It has in vitro activity against a range of gram-positive and some gram-negative bacteria [4]. Mupirocin has excellent activity against staphylococci and gram-negative organism such as Haemophilus influenza, Neisseria gonorrhoeae and Moraxella catarrhalis [5]. Drug is available in market in the form of ointment and cream. HPLC method for the measurement of Mupirocin concentrations in both skin layers and percutaneous samples has been reported [6]. A simple UV-Spectroscopic method for estimation of Mupirocin calcium from ointment formulation has been reported [7]. HPTLC method for estimation of Mupirocin from ointment formulation has been reported [8]. Analysis of pharmaceutical parameters is an imperative and chief aspect of the complete drug development process. Therefore, simple and rapid methods for quality control of commercial formulations are needed.

Chemical structure of Mupirocin calcium
Forced degradation experiments are used to relieve the development of analytical methodology, to achieve better insightful of the stability of the active pharmaceutical ingredient (API) and the drug product, and to provide information about degradation pathways and degradation products [9]. However, no literature is available for which deals with the stress degradation profile of Mupirocin calcium in accordance with ICH guidelines using any of the above analytical techniques. The table below depicts existing literature disadvantages and reported method advantages:
No research data have been found for estimation of MUP by HPLC in dosage form. The objective of this work was to develop a stability-indicating reversed-phase high-performance liquid chromatography (RP-HPLC) for analysis of MUP in pharmaceutical formulation. This paper describes an accurate, specific, repeatable, and stability-indicating method for analysis of Mupirocin calcium in the presence of its degradation products. The method was validated in accordance with the guidelines of International Conference on Harmonization (ICH) [10].
Necessity and importance of stability-indicating method
The goal of the stabilization studies is to track potential improvements to a substance or material over time and under various storage conditions. The factors and parameters that affect the stability are production timeframe, batch factors along with process parameters, excipients efficiency, and environmental conditions like temperature and humidity [11, 12].
The access to appropriate deteriorated samples for method production assistance is a major challenge when designing a stability indicator method (SIM). Such deteriorated samples in a perfect environment must be real-time stability samples containing all applicable degradant as well as those degradant develop during ordinary storage conditions. For this cause, pharmacists must use forced degradation samples to create SIMs. Many experiments have explored the potential of forced deterioration studies to predict real-time degradation [13].
The precision of the stability methods showing potential impurities of the drug material and of drug components is demonstrated by forced degradation (FD). Stress experiments help to generate impurities in a much shorter period. The formulations scientist will then generate consistent formulations in less time. FD studies now include the completion of the file and the comprehension of the drug production mechanism for global controlled markets [14].
GMP includes a structured written monitoring program for stability, the results of which can be used to specify the storage requirements, the expiry dates and the use of accurate, meaningful and precise test procedures. If there is an effort to document drug product stability, the use of such approaches is acceptable. These data are being used to assess, conform or expand retest cycles or expiration date for the drug substance [15].
The rationale for the stability studies research is to provide data as to how the consistency of the substance varies over the time under the control of a multiplicity of ecological variables, such as humidity, temperature and light, allows the proposed storage conditions, re-analysis periods and shelf life [16, 17].
Methods
Reagents and chemicals
Mupirocin calcium was supplied as a gift sample by Glenmark Pharmaceuticals Ltd., Mumbai, India. All the chemicals used of HPLC grade (MERCK Chem. Ltd., Mumbai) and double distilled R.O. water were used for mobile phase preparation. Solvents and solutions were filtered through a membrane filter (0.45 μm pore size) and degassed by sonication before use.
Instrumentation
The chromatographic analysis was performed on Shimadzu HPLC system equipped with PDA detector. The output signals were monitored and processed using LC Solution software. The analytical column was PrincetoneSPHER-100 C8 (4.6 mm × 250 mm, 5 µ) and the samples were introduced through a Rheodyne injection valve with 20 μL sample loop.
Wavelength detection
MUP was accurately weighed 10 mg and transferred in 100 mL volumetric flask, and methanol was added up to the mark. From that solution, 1.2 mL was pipetted out and diluted with methanol in 10 mL volumetric flask. The scanning was done between 200 and 400 nm by UV spectroscopy.
Chromatographic conditions
In order to get sharp peak, various proportions for mobile phase consisting of methanol and water were tried, to get adequate resolution of MUP. Initially, methanol: water (80: 20% v/v) was attempted; it gave Rt at 7.36 min with tailing. Further modification was done by taking methanol: water (70:30 % v/v) and pH adjusted to 4 with acetic acid which resolve it gave Rt at 4.61 min with tailing. In order to have well resolved a sharp peak, finally selected mobile phase was methanol and water (pH adjusted to 4 with acetic acid) in the ratio 75:25 v/v. Injection volume was 20 μL, flow rate was 1.0 mL/min and the eluent was detected at 221 nm at column temperature 30 °C. These conditions showed sharp peak of MUP with retention time of 5.09 min (Fig. 2).

Sharp peak of Mupirocin calcium in mobile phase methanol: water (75: 25) pH adjusted to 4 with acetic acid at 221 nm with RT 5.09 min
Preparation of stock standard solution and sample
Standard stock solution was prepared by dissolving 10 mg of MUP in 100 mL methanol that gives concentration of 100 μg/mL. This solution was diluted with mobile phase as needed to prepare different standard solutions.
Analytical method validation
Determination of linearity
From the stock solution, aliquots of 0.4, 0.8, 1.2, 1.6, 2.0 and 2.4 mL were taken in 10 mL volumetric flasks and diluted up to the mark with mobile phase to get the final concentration in range of 4–24 μg/mL. Calibration curve was constructed by plotting the peak area vs. the drug concentration (Fig. 3).

Calibration curve of Mupirocin calcium. Y = 30420x + 66435Slope = 30420, Intercept = 66435, Correlation Coefficient = 0.9998
Precision
Precision can be performed at two different levels: repeatability and intermediate precision. Repeatability refers to the use of the analytical procedure within the laboratory over the shorter period of the time that was evaluated by assaying the samples during the same day. Repeatability was carried out using six replicates of the sample injection. Intra-day precision was determined by analyzing, the three different concentrations 8, 12 and 16 μg/mL of MUP, for three times in the same day. Day to day variability was assessed using above mentioned three concentrations analyzed on three consecutive days for inter-day precision.
Accuracy
Accuracy was done by recovery study using standard addition method at 80%, 100% and 120% level; known amount of standard MUP was added to the sample and subjected to the proposed HPLC method. The accuracy studies were carried out three times, and the % recovery and % RSD were calculated.
Precision and accuracy study were performed as per the ICH guidelines. For precision study, three different concentrations 8, 12 and 16 μg/mL of MUP were taken. While for accuracy, the standard addition method at 80%, 100% and 120% level is used.
Limit of detection (LOD) and limit of quantitation (LOQ)
Sensitivity of the proposed method was estimated in terms of LOD and LOQ. LOD is the lowest concentration in a sample that can be detected, but not necessarily quantified; under the stated experimental conditions. LOQ is the lowest concentration of analyte in a sample that can be determined with acceptable precision. In order to determine LOD and LOQ, Mupirocin calcium concentrations in the lower part of the linear range of the calibration curve were used. For this, very dilutions viz. 4.5, 5, 5.5, 6, 6.5, 7µg/mL were prepared for analysis. Limit of quantitation (LOQ) were calculated by the use of equation, LOQ = SD/S × 10, where SD is the residual standard deviation of the peak areas of the drugs (n = 6) and ‘S’ is the slope of the line. Sensitivity was performed between 4–8 µg/mL, (slope- 30,420, S/N ratio = 10/1)
Analysis of marketed formulation
Commercially available formulation of MUP (Bactroban cream) (GlaxoSmithKline ltd.) was selected for estimation of total drug content by proposed method. An amount of formulation equivalent to 0.2 g of Mupirocin calcium was weighed accurately and transfer to 100 mL volumetric flask. Add 50 mL mobile phase and sonicated for 10 min. Volume was adjusted to 100 mL with the mobile phase. The resulting solution was filtered using 0.45 μm filter and further diluted for analysis to get a concentration of 12 µg/mL. The formulation consists of oil and water soluble base, so it is highly soluble in methanol and water solvent.
Robustness
Robustness of the method was studied by making small deliberate changes in few parameters. The flow rate and temperature were varied by ± 0.2 mL/min and ± 1 °C, respectively. The flow rate (1 ± 0.2 mL i.e. 0.8, 1.2) and temperature (30 ± 1 °C i.e. 29, 31) were varied. Robustness was assessed at concentration of 12 µg/mL.
Ruggedness
From stock solution, sample solution of MUP (12µg/mL) was prepared and analyzed by two different analysts using similar operational and environmental conditions. Peak area was measured for same concentration solutions, six times.
System suitability test
System suitability testing is essential for the assurance of the quality performance of chromatographic system. Earlier prepared solutions for chromatographic conditions were tested for system suitability testing.
Forced degradation studies
Acid- and base-induced degradation was attempted by separately adding 10 mg of drug in 50 mL each of methanolic 0.1 N HCl and methanolic 0.1 N NaOH solutions separately. These solutions were kept for 6h at room temperature in the dark in order to exclude the possible degradative effect of light. The solutions (1 mL) were neutralized and diluted to 10 mL with methanol. The 20 µL (20 µg/mL) of the resulting solutions was injected and analyzed. Since no degradation has observed at room temperature, further the temperature is increased. Acid- and base-induced degradation was attempted by separately adding 10 mg of MUP in 50 mL each of methanolic 0.1 N HCl and methanolic 0.1 N NaOH solution separately.
The resulting solutions were refluxed at 60 °C for 6 h. 1 mL sample was withdrawn at different intervals for 6 h which was neutralized and diluted to 10 mL with methanol. The 20 µL of the resulting solutions (20 µg/mL) was injected and analyzed.
For oxidative degradation, 10 mg of MUP was added to 50 mL of methanolic 5% (v/v) hydrogen peroxide solution and was kept in the dark for 6h. The solution (1 mL) was diluted to 10 mL with methanol and treated as described for acid- and base-induced degradation. No degradation has occurred. Further, the oxidative degradation has extended by taking 10 mg of Mupirocin calcium, added in 50 mL of methanolic 10% (v/v) hydrogen peroxide solution, and resulting solution was kept for 24 h. The solution (1 mL) was withdrawn and diluted to 10 mL with methanol. The 20 µL of resulting solutions (20 µg/mL) was injected and analyzed.
For photodegradation, drug was kept under sunlight for 12 h, and then, 10 mg of Mupirocin calcium added in 50 mL of methanol. The solution (1 mL) was withdrawn and diluted to 10 mL with methanol. The 20 µL of resulting solution (20 µg/mL) was injected and analyzed.
Results
To develop an accurate, precise and specific stability-indicating RP-HPLC method for estimation of MUP using stressed samples, various mobile phases with different composition and flow rate were tried. After several compositions and permutations, chromatographic conditions were optimized and established. Satisfactory estimation of MUP with good peak symmetry and steady baseline was obtained with the mobile phase methanol: water (pH adjusted to 4 using acetic acid) in the ratio 75:25 v/v at a flow rate of 1.0 mL/min. Drug showed single sharp peak at retention time (RT) of 5.09 min with clear baseline at 221 nm. The detailed validation parameters are summarized in Table 1.
Linearity
The standard curve for Mupirocin calcium was linear over the investigated concentration range 4–24 µg/mL with a percent relative standard deviation (% RSD) of not more than 2 based on five successive readings. The linearity equation was obtained with a slope of 30420, intercept of 66435 and a correlation coefficient (r2) of 0.9998.
Precision
The precision of an analytical method is the degree of agreement among individual test results obtained when the method is applied to multiple sampling of a homogenous sample. Precision studies of proposed method were determined by repeatability and intermediate precision (intra-day and inter-day precision). Repeatability was measured by multiple injections of 12 µg/mL of MUP that indicates the performance of the HPLC instrument under chromatographic conditions. The % RSD was found to be within the limit indicating the proposed method is more precise as listed in Table 2.
Recovery
The mean recovery data of MUP in sample were 99.61%, while % RSD was 0.33, that satisfying the acceptance criteria for the study. It proved that there is no interference of excipients used in formulation as seen by results tabulated in Table 3.
Limit of detection (LOD) and limit of quantitation (LOQ)
The LOD with signal-to-noise (S/N) ratio of 3:1 and the LOQ with S/N ratio of 10:1 were calculated for MUP using the equations LOD = 3.3 × N/B and LOQ = 10 × N/B where ‘N’ is the standard deviation of the peak areas of the drug (n = 3), taken as measure of the noise, and ‘B’ is the slope of corresponding calibration plot. The signal-to-noise ratio was determined. LOD and LOQ were found to be 0.35 and 1.08 µg/mL, respectively.
Assay
By taking the mean of six determinations, the amount found for MUP was 99.26%. From the data obtained, %RSD of drug was found to be within the limits. Thus, it can be concluded that there is no interference of the excipients as depicted in Table 4.
Robustness
In robustness studies, small and deliberate changes in flow rate and temperature were made. With respect to this, change in retention time and tailing factor was observed. The flow rate and temperature were varied by ± 0.2 mL/min (i.e., 0.8 & 1.2 mL/min) and ± 1 °C (i.e., 29 & 31 °C), respectively. Robustness was assessed at concentration of 12 µg/mL. The standard deviation of response was calculated for each parameter and % RSD was found to be less than 2% indicating that the method is robust as shown in Table 5.
Ruggedness
Ruggedness of the method was studied by two different analysts. Method proved to be rugged as it showed low values of % RSD.
System suitability test
The number of theoretical plates and other system suitability parameters were calculated. They were found to be within limits as listed in Table 6.
Degradation studies
The chromatograms obtained from samples exposed to acidic, alkaline, oxidative and photodegradation depicted well-separated peaks of pure MUP having Rt 5.09 min and some additional peaks at different values. Acidic and basic degradation shows 3 additional peaks for each. In case of oxidative degradation shows 1 additional peak. In case of photo degradation shows 1 additional peak. The % of degradation products with their Rt values is listed in Table 7. Results of Force degradation are shown in Figs. 4, 5, 6, 7.

Acid degradation in which Mupirocin calcium undergoes degradation in presence of methanolic 0.1 N HCl at 60 °C

Base degradation in which Mupirocin calcium undergoes degradation in presence of methanolic 0.1 N NaOH at 60 °C

Oxidative degradation in which Mupirocin calcium undergoes degradation in presence of methanolic 10% H2O2 at RT

Photodegradation in which Mupirocin calcium undergoes degradation under sunlight for 12 h
Discussion
In this work, an effective resolution of Mupirocin calcium was successfully performed and developed using the HPLC technique. Thus, the proposed study had various advantages over the existing one. In existing method, the solvent used was Acetonitrile which is expensive; the mobile phase composition was a mixture of acetonitrile-ammonium acetate, while the proposed method seems to be economic as it consumes methanol as solvent along with use of water in mobile phase. The retention time was less 5.09 ± 0.02 min at 221nm wavelength as compared to existing method having is7.20 ± 0.07 min. Also, there is promising difference in LOD and LOQ values with 9.5 ng/mL and 30.6 ng/mL for existing method and 0.35 μg/mL and 1.08 μg/mL for proposed one. The peak tailing is 1.32 against 1.5. Forced degradation study was performed by applying various stress conditions to the sample to evaluate the robustness and stability-indicating nature of the developed method. We detected that degradations of Mupirocin calcium were more prominent in acidic and basic environments within the forced degradation experiment. Mupirocin Calcium did not degrade further when exposed to oxidative conditions and light. The peak size, theoretical plates, and tailing factor for peak were determined to be within the acceptable limits during the robustness analysis, confirming the method's robustness. The comparison of existing and proposed method is tabulated in Table 8 (Fig. 8).

Peak purity of Mupirocin
Conclusions
The modalities adopted in experiment were successfully validated as per ICH guidelines; analytical procedures laid down in routine analysis. The proposed method was validated by preliminary analysis of standard sample and by recovery studies.
A validated stability-indicating RP-HPLC method has been developed for the determination of Mupirocin calcium in bulk and in pharmaceutical dosage form. The proposed method is found to be simple, rapid, accurate and precise. The statistical evaluation of the proposed method was revealed its good linearity and its validation for different parameters and let us to the conclusion that it could be used for the rapid and reliable determination of Mupirocin calcium in pharmaceutical formulation. From the results and discussion, we can conclude that the method is suitable for estimation of Mupirocin calcium and under stress conditions the drug undergoes possible degradation.
Availability of data and materials
Not applicable.
Abbreviations
- API:
-
Active pharmaceutical ingredients
- FD:
-
Forced degradation
- GMP:
-
Good manufacturing practice
- HCl:
-
Hydrogen chloride
- HPLC:
-
Reversed-phase high-performance liquid chromatography
- HPTLC:
-
High-performance thin-layer chromatography
- ICH:
-
International Conference on Harmonization
- LOD:
-
Limit of detection
- LOQ:
-
Limit of quantitation
- MUP:
-
Mupirocin calcium
- PDA:
-
Parenteral Drug Association
- RNA:
-
Ribonucleic acid
- RP-HPLC:
-
Reversed-phase high-performance liquid chromatography
- RSD:
-
Relative standard deviation
- RO:
-
Reverse osmosis
- SIM:
-
Stability indicator method
- UV:
-
Ultraviolet
References
British Pharmacopoeia (2004) Government of British, Monograph for Mupirocin calcium
Hughes J, Mellows G (1978) Inhibition of isoleucyl-transfer ribonucleic acid synthetase in Escherichia coli by psedomonic acid. Biochem J 176(1):305–318
Hughes J, Mellows G (1980) Interaction of pseudomonic acid A with Escherichia coli B of isoleucyl-tRNA synthetase. Biochem J 191(1):209–219
Vizcaino-Alcaide MJ, Herruzo-Cabrera R, Rey-Calero J (1993) Efficacy of broad spectrum antibiotic (Mupirocin) in an in vitro model of infected skin. Burns 19(5):392–395
Sutherland R, Boon RJ, Griffin KE, Masters PJ, Slocombe B, White AR (1985) Antibacterial activity of Mupirocin (pseudomonic acid) a new antibiotic for topical use. Antimicrob Agents Chemother 27(4):495–498
Echevarria L, Blanco Prieto MJ, Campanero MA, Santoyo S, Ygartuay P (2003) Development and validation of a liquid chromatographic method for in vitro Mupirocin quantification in both skin layers and percutaneous penetration studies. J Chromatogr B 796(2):233–241
Bageshwar DV, Pawar A, Khanvilkar V, Kadam VJ (2010) Quantitative estimation of mupirocin calcium from pharmaceutical ointment formulation by UV spectrophotometry. Int J Pharm Pharm Sci 2:86–88
Chhajed SV, Potawale SE, Jagdale SR, Gabhe SY, Mahadik KR (2013) Densitometric development and validation of Mupirocin in ointment dosage form Pelagia Research Library. Der Pharm Sin 4(6):10–15
Alsante KM, Ando A, Brown R, Ensing J, Hatajik TD, Kong W et al (2007) The role of degradant profiling in active pharmaceutical ingredients and drug products. Adv Drug Del Rev 59:29–37
ICH-Q2B (2005) Guidelines for industry, validation of analytical procedures: methodology. In: International conference on harmonization IFPMA Geneva.
Aubry AF, Tattersall P, Ruan J (2009) Development of stability indicating methods. In: Huynh-Ba K (eds) Handbook of stability testing in pharmaceutical development. Springer, New York. https://doi.org/10.1007/978-0-387-85627-8_7
Dongala T, Katari NK, Palakurthi AK, Katakam LNR, Marisetti VM (2020) Stability indicating LC method development for hydroxychloroquine sulfate impurities as available for treatment of COVID-19 and evaluation of risk assessment prior to method validation by quality by design approach. Chromatographia 83:1269–1281
Maheswaran R (2012) FDA perspectives: scientific considerations of forced degradation studies in ANDA submissions. Pharm Technol 36:73–80
ICH guidelines: Q1A (R2) (2003) Stability testing of new drug sub-stances and products (revision 2). In: International conference on harmonization
Reynolds DW, Facchine KL, Mullaney JF (2002) Available guidance and best practices for conducting forced degradation studies. Pharm Technol 26:48–56
Blessy M, Patel D, Prajapati P (2014) Development of forced degradation and stability indicating studies of drugs—a review. J Pharm Anal 4:159–165
Soni NR (2018) Stability indicating analytical methods (SIAMS)
Acknowledgements
Authors are thankful to Glenmark pharmaceuticals Ltd., Mumbai (India), for providing gift samples of Mupirocin calcium.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
DJK contributed for experimental work and manuscript preparation. VKR contributed in hypothesis and finalization of manuscript. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kalal, D.J., Redasani, V.K. Stability-indicating RP-HPLC method development and validation for estimation of Mupirocin calcium in bulk and in pharmaceutical formulation. Futur J Pharm Sci 8, 21 (2022). https://doi.org/10.1186/s43094-022-00412-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43094-022-00412-w