
Abstract
Background
A simple and sensitive gas chromatographic method was developed and validated for the simultaneous determination of methanol, ethanol, acetone, isopropyl alcohol, dichloromethane, N-hexane, ethyl acetate, tetrahydrofuran, and N,N-diisopropyl ethyl amine in Paclitaxel. A chromatographic separation was done on DB-624 column, 30 m length × 0.53 mm ID, and film thickness 3 μm, using a flame ionization detector (FID) with gradient column oven temperature program. The injection was carried out in split mode, with a split ratio of 5:1. A mixture of N-methyl-2-pyrrolidinone (contains 1% piperazine) and water in the ratio of 80:20 (v/v) was selected as a diluent to obtain good sensitivity along with the recovery.
Results
The developed gas chromatographic method offers symmetric peak shape, good resolution of more than 2.0 between the solvent peaks, and the relative standard deviation for replicate injections of all the solvents were found to be not more than 15.0% with reasonable retention time for all the solvents. The limit of detection for methanol, ethanol, acetone, isopropyl alcohol, dichloromethane, N-hexane, ethyl acetate, tetrahydrofuran, and N,N-diisopropyl ethyl amine was found to be 304.69 ppm, 497.98 ppm, 498.99 ppm, 504.49 ppm, 61.81 ppm, 30.07 ppm, 505 ppm, 73.05 ppm, and 2.09 ppm, respectively. Limit of quantitation of methanol, ethanol, acetone, isopropyl alcohol, dichloromethane, N-hexane, ethyl acetate, tetrahydrofuran, and N,N-diisopropyl ethyl amine was found to be 89.62 ppm, 146.47 ppm, 146.76 ppm, 148.38 ppm, 18.18 ppm, 8.84 ppm, 148.53 ppm, 21.49 ppm, and 0.62 ppm, respectively. Precision was found to be satisfactory. Linear in the range of LOQ to 150% level for all the solvents, and accuracy along with robustness, is performed, and acceptable results were obtained.
Conclusion
The proposed method was demonstrated to be simple, sensitive, specific, linear, precise, accurate, and robust, hence can be used to determine the residual organic solvents in Paclitaxel drug substance and drug product.
Similar content being viewed by others

Background
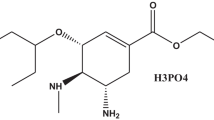
Paclitaxel [1] (Fig. 1) is a taxane derivative originally derived in limited amounts from the bark of the pacific yew tree Taxus brevifolia (Taxaceae). It is now obtained from a taxane precursor derived from the needles of the European yew, Taxus bacata, using a semi-synthetic process. It is a BCS class IV drug with a high degree of hydrophobicity and consequently an extremely low aqueous solubility of 4 μg/mL [2, 3]. Paclitaxel has shown significant activity against a wide range of tumors such as those in breast, ovarian, and lung cancer, in addition to head and neck carcinomas [4]. An impurity in a drug substance was defined by the International Conference on Harmonisation (ICH) guidelines that are any component of the drug substance that is not the chemical entity defined as the drug substance and affects the purity of active ingredient or drug substances [5]. Similarly, an impurity in a drug product is any component of the drug product that is not the chemical entity defined as the drug substance or an excipient in the drug product [6]. Therefore, any extraneous material present in the drug substance has to be considered an impurity even if it is inert or has superior pharmacological properties. The impurity profile of pharmaceuticals is of increasing importance as drug safety receives more and more attention from the public and the media. Several recent books and journal reviews address this topic, and guidelines are available from the USA and international authorities [7,8,9,10,11,12,13,14]. Most active pharmaceutical ingredients (APIs) are produced by organic chemical synthesis. Various components including residual solvents trace amounts of inorganic, and organic components, can be generated during such processes.

Chemical Structure of Paclitaxel. Molecular Formula: C47H51NO14. Molecular weight: 853.9 g/mol
Analysis of a residual solvent in pharmaceuticals is an important issue due to the potential risk to human health from the toxicity of many of these solvents. The amount of such solvents is, therefore, limited by ICH guidelines [15]. The ICH has published the limits of the residual solvents that considered safe in pharmaceutical preparations; also, it has published the daily exposure limits for these solvents. It has classified these solvents in three categories depending on their toxicity. Class I solvents are known human carcinogens and environmental hazards, and the use of these solvents should be avoided if at all possible. Class II solvents are non-genotoxic animal carcinogens or possible causative agents of other irreversible toxicities such as neurotoxicity or teratogenicity. The use of these solvents should be limited. Class III solvents are the solvents with the low toxic potential to man, and no health-based exposure limit is needed. The list of solvents with their class and limits is given in Table 1. In the pharmaceutical industries, all the pharmaceutical products must be analyzed for residual solvent content, regardless of the matrix.
Gas chromatography is generally used to determine residual solvents due to its excellent separation ability and high sensitivity. In gas chromatography, the sample is either dissolved in a suitable solvent than injected directly [16] or by headspace sampling. Headspace sampling is preferred due to its ability to avoid direct liquid or solid probing. In the headspace sampling, complex sample matrix in a solid or liquid sample matrix in the liquid or solid sample can be simplified or even eliminated in its vapor phase [17]. Different methods have been reported in the literature for the determination of Paclitaxel, e.g., capillary electrophoresis [18], LC-MS [19], and high performance liquid chromatography (HPLC) [20, 21]. Also, there are many reports that use HPLC technique to determine related substances in plant extracts, raw material, and taxol preparations [22,23,24,25,26,27,28,29].
The objective of this work is to develop and validate a new gas chromatographic method for the simultaneous determination of residual solvents in Paclitaxel. These solvents should be estimated and checked so that they may not exceed the amount specified by the ICH guidelines.
Methods
Materials
Paclitaxel raw material was procured from the Spectrum Pharma Research Private Limited, Hyderabad, India. GC grade methanol, ethanol, acetone, isopropyl alcohol, dichloromethane, n-hexane, ethylacetate, tetrahydrofuran, N,N-diisopropyl ethylamine, N-methyl 1-2-pyrrolidinone, and piperazine were purchased from the Merck India Limited, Mumbai, India.
Instrumentation
A gas chromatograph (Agilent Technologies 6890A) equipped with flame ionization detector (FID) connected to Agilent G1888 Headspace sampler and a data processor Waters Empower 3 software was employed. The column utilized was DB-624, 30 m length × 0.53 mm ID, and film thickness 3 μm. Meltronics sonicator was used to enhance the solubility of the material. Sartorius balance was employed for weighing the samples.
Optimized chromatographic conditions
Various GC columns such as DB-1 and DB-5 were used of various dimensions, but the best separation was achieved on DB-624, 30 m length × 0.53 mm ID, and film thickness 3 μm. Details of other optimized gas chromatographic and headspace parameters are given in Tables 2 and 3, respectively.
Preparation of diluent (mixture of N-methyl-2-pyrrolidinone (contains 1% piperazine and water in the ratio of 80:20 v/v)
Accurately weigh and transfer about 1.0 g of piperazine in 100 mL volumetric flask (1%). Add about 25 mL of N-methyl-2-pyrrolidinone (NMP) solvent to this volumetric flask. Sonicate the flask till the piperazine completely dissolves in N-methyl-2-pyrrolidinone (NMP). Transfer 20 mL of water into this solution (20%). Mix this solution thoroughly and adjust to volume with the same solvent N-methyl-2-pyrrolidinone.
Blank solution
Transfer 1 mL of diluent into Agilent Technologies manufactured 20 mL flat bottom headspace GC vials fitted with a septum and crimp cap and seal. The chromatogram of blank solution showed in Fig. 2.

Typical Chromatogram of Blank
Preparation of N,N-diisopropyl ethyl amine stock solution (DIPEA)
Accurately weigh and transfer about 20 mg of DIPEA in 10 mL volumetric flask containing about 5 mL of diluent and mix and adjust to volume with diluent.
Preparation of DIPEA standard solution
Transfer 1 mL of above DIPEA standard stock solution into 20 mL volumetric flask containing 5 mL of diluent and mix and adjust to volume with diluent.
Preparation of standard stock solution-A
Accurately weigh and transfer about 150 mg of methanol, 250 mg of ethanol, 250 mg of acetone, 250 mg of isopropyl alcohol, 250 mg of ethyl acetate, 36 mg of tetrahydrofuran, 30 mg of dichloromethane, and 15 mg of n-hexane in 10 mL volumetric flask containing about 1 mL of diluent mix and made up to the mark with diluent.
Preparation of standard solution
Transfer 0.8 mL of DIPEA standard solution and 0.8 mL of standard stock solution-A into 50 mL volumetric flask containing about 20 mL of diluent and mix until the volume was made up to the mark with diluent.
Transfer 1 mL of this solution into 20 mL headspace GC vial and seal vial adequately fitted with a septum and crimp cap.
This standard solution contains about 3000 ppm of methanol, 5000 ppm of ethanol, 5000 ppm of acetone, 5000 ppm of isopropylalcohol, 5000 ppm of ethylacetate, 720 ppm of tetrahydrofuran, 600 ppm of dichloromethane, 290 ppm of n-hexane, and 20 ppm of DIPEA (with respect to test concentration). The chromatogram of standard solution showed in Fig. 3.

Typical Chromatogram of Standard
Sample preparation
Weigh accurately about 80 mg of sample for evaluation into 20 mL flat bottom headspace GC vials and add 1 mL of diluent fitted with a septum and crimp cap and seal.
Results
Method validation
The developed method was validated according to the ICH guidelines with reference to accuracy, precision, system suitability, specificity, linearity, limit of quantification, limit of detection, and robustness [30].
System suitability
System performance parameters of the optimized GC method were determined by analyzing standard solution. Chromatographic parameters such as number of theoretical plates, tailing factor, and resolution were determined. The results are within the specifications, indicating the excellent performance of the system. System repeatability was established by six replicate injections of the standard solution, and the relative standard deviations (RSD) for the peak area of the solvents were calculated to evaluate the repeatability. The obtained results were within the ICH permissible limits mentioned in Table 4. The blank chromatogram is shown in Fig. 2, and the typical chromatogram shows that all the solvents are shown in Fig. 3.
Linearity
The linearity of the relationship between the peak area and the concentration in ppm evaluated for all the residual solvents mentioned in the present study was investigated by linear regression analysis. Six linearity solutions were prepared to range from limit of quantitation LOQ to 150% of the specified level concentration of each solvent. The linear range investigated for each solvent is mentioned in Tables 5, 6 and 7. Linearity curves were drawn by plotting the graph of the average peak area of solvent against its concentration in ppm for linearity solutions, Figs. 4, 5, 6, 7, 8, 9, 10, 11, and 12.

Linearity plot of Methanol

Linearity plot of Ethanol

Linearity plot of Acetone

Linearity plot of Isopropyl Alcohol

Linearity plot of Dichloromethane

Linearity plot of n-Hexane

Linearity plot of Ethylacetate

Linearity plot of Tetrahydrofuran

Linearity plot of DIPEA
Accuracy and precision
Both the terms accuracy and precision are mutually correlated, where accuracy is the difference between the true value and the observed value. With the precision, it has a limited significance. Accuracy and precision were determined by applying the optimized method in which known amount of each solvent corresponding to LOQ, 50%, 100%, and 150% of specified target concentration. Each level was prepared in triplicate. The accuracy was then calculated as the percentage of analyte recovered. From the results, it is evident that the recovery of each in spiked samples ranged from 97.0 to 115.0%. Mean recoveries for all the solvents are shown in Tables 8, 9, 10, 11, 12, 13, 14, 15 and 16.
The precision of an analytical procedure expresses the closeness of agreement (degree of scattering) between a series of measurements obtained from multiple sampling of the same homogeneous sample under the prescribed conditions. Precision may be considered at three levels: repeatability, intermediate precision, and reproducibility. The precision of an analytical procedure is usually expressed as the variance, standard deviation, or coefficient of variation of a series of measurements. Method precision shall be established by determining the assay in six different preparations of a standard solution. Intermediate precision shall be determined by studying the variation in assay of a homogeneous sample analyzed by two different equipment, analyst and days. The average, standard deviation, and relative standard deviation shall be calculated. The results for the method and intermediate precision are found to be under the acceptable limit for each residual solvent as revealed by relative standard deviation data (RSD < 15.0% for the solvents). The precision results are shown in Table 17.
Limit of detection (LOD) and limit of quantitation (LOQ)
The limit of detection of an individual analytical procedure is the lowest amount of analyte in a sample, which can be detected but not necessarily quantitated as an exact amount. While the limit of quantitation was the minimum level of concentration of analyte at which it can be quantitated with acceptable precision and accuracy. LOD and LOQ were calculated using the signal-to-noise ratio (S/N) method using the Empower software. Six replicate solutions were injected into the chromatograph and recorded. Obtained LOD and LOQ of each solvent are mentioned in Table 18.
Robustness
For robustness, three deliberate changes were done concerning carrier gas flow rate, column oven temperature, and vial oven temperature. Each change consists of one upper set and one lower set. For each set, six replicate determinations were analyzed. The results were found to be satisfactory and within the acceptable limits. The obtained results are mentioned in Tables 19, 20, 21, 22, 23, 24, 25, 26, and 27.
Discussion
In this research study involved a new gas chromatographic method for determination of residual solvents in bulk. Different methods have shown by using different analytical techniques for the determination of Paclitaxel [18,19,20,21,22,23,24,25,26,27,28,29]. However, the gas chromatographic method for determination of residual solvents has not been reported earlier.
The primary goal of this study is to provide a simple and sensitive gas chromatographic method for the determination of all the residual solvents present in the active analyte. During the development of the analytical method, trails were done and optimized the method and found to be feasible and can be adoptable. The system suitability parameters like injection repeatability, number of theoretical plates, tailing factor, and resolution results were met the USP acceptance limits (Table 4), which resembles integrity of the system.
The retention time of the solvent peaks of standard solution matches with that of the spiked test sample solution. No interference was observed at a retention time of the solvent peak from blank and test sample Figs. 2 and 3 which clearly resembles the specificity of the proposed method. The percentage recovery obtained for each solvent was in the range of 80–120%, which is within ICH acceptance. Precision parameter shows that the RSD was < 5.0% for all the solvents in system precision, repeatability, and intermediate precision at 100% concentration which proved that the developed analytical method was accurate and precise (Tables 8, 9, 10, 11, 12, 13, 14, 15, 16 and 17). Linearity was observed in the concentration range of LOQ to 150% with r2 values > 0.999 and y-intercept < 5.0% showing a good correlation between the response and solvent concentration (Tables 5, 6 and 7). The linearity of the method was confirmed statistically. The calculated limit of detection and limit of quantitation for each solvent found to be satisfactory (Table 18). The method is robust as in robustness parameter with deliberate changes made for which individual and cumulative RSD values for each set were < 5.0% (Tables 19, 20, 21, 22, 23, 24, 25, 26, and 27). All the obtained results from the validation parameters were found to be meeting to the ICH acceptance criteria [30]. Finally, the anticipated method was found to be suitable for the routine analysis in the research laboratories as well as in the quality control.
Conclusion
The developed gas chromatographic method with FID detector offers simplicity, selectivity, precision, accuracy, and robust. It produces symmetric peak shape and reasonable retention time for all the solvents. It often can be seen from the chromatogram that all the solvents were eluted before 25 min of injection of sample. It can be used for the determination of residual solvents in PACLITAXEL API, and this method can even be used to separate the residual solvents present in other drug substances and also within the finished dosage forms where the particular solvents used for the coating purpose or any other excipients within the pharmaceutical companies and research laboratories and also be advantageous for scale manufacturing purpose.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- API:
-
Active pharmaceutical ingredient
- DIPEA:
-
N,N-di-isopropyl ethyl amine
- FID:
-
Flame ionization detector
- ICH:
-
International Conference on Harmonisation
- NA:
-
Not applicable
- NLT:
-
Not less than
- NMT:
-
Not more than
- NMP:
-
N-methyl-2-pyrrolidinone
- RSD:
-
Relative standard deviation
- S/N:
-
Signal-to-noise ratio
References
O’Neil MJ, Smith A, Heckelman PE (2006) The Merck Index, An Encyclopedia of Chemicals, Drugs and Biologicals, 14th edn. Merck & Co. Inc., White House Station, p 1204
Sweetman SC (2006) Martindale the Complete Drug Reference, 35th edn. Pharmaceutical Press, London, p 685
Zentner GM, Rathi R, Shih C, McRea JC, Seo M, Rhee BG, Mestecky J, Moldoveanu Z, Morgan M, Weitman S (2001) Biodegradable block copolymers for delivery of proteins and water-insoluble drugs. J Control Release 72:203–215
Arbuck SG, Blaylock B (1995) TAXOL: clinical results and current issues in development. In TAXOL: Science and Applications. M. Suffness, ed. CRC Press, New York, p 379
U.S. Food and Drug Administration (2003) Q3A Impurities in New Drug Substances. Food and Drug Administration, Washington, DC
U.S. Food and Drug Administration (2006) Q3B Impurities in New Drug Products. Food and Drug Administration, United States
Gorog S (2000) Identification and Determination of Impurities in Drugs. Elsevier Science, B.V, Amsterdam
Bhowmik H, Venkatesh DN (2001) Nanosponges: A review. Int J Appl Pharms 10:1–5
Hovorka S, Schöneich C (2001) Oxidative degradation of pharmaceuticals: Theory, mechanisms and inhibition. J Pharm Sci. 90:253–269
Roy J (2002) Pharmaceutical impurities – a mini-review. AAPS Pharm Sci Tech 3:E6
ICH Harmonised Tripartite Guidelines: Q3A(R2). Impurities in New Drug Substances International Conference on Harmonisation; 2006
ICH Harmonised Tripartite Guidelines: Q3B(R6). Impurities in New Drug Products International Conference on Harmonisation; 2006
ICH Harmonised Tripartite Guidelines: Q3C(R2). Maintenance of the guidelines for Residual Solvents; International Conference on Harmonisation; 2016
ICH Harmonised Tripartite Guidelines: Q1A(R2). Stability Testing of New Drug Substances and Products; International Conference on Harmonisation; 2003
Haky JE, Stickney TM (1985) Automated gas chromatographic method for the determination of residual solvents in bulk pharmaceuticals. J Chrom. 321:137–144
Markovich RJ, Ong S, Rosen J (1997) J Chrom Sci 35:584–592
Zhu JY, Chai XS (2005) Some recent developments in headspace gas chromatography. Curr Anal Chem. 1:79–83
Hempel G, Lehmkuhl D, Krumpelmann S, Blaschkle G, Boos J (1996) Determination of paclitaxel in biological fluids by micellar electrokinetic chromatography. J Chrom A. 745:173
Theodoridis G, Laskaris G, Rozendaal ELM, Verpoorte R (2001) Analysis of taxines in Taxus plant material and cell cultures by HPLC photodiode array and HPLC-electrospray mass spectrometry. J Liquid Chrom. 24:2267
Nguyen T, Eshraghi J, Gonyea R, Ream R, Smith R (2001) Studies on factors influencing stability and recovery of paclitaxel from suspension media and cultures of Taxus cuspidata Densiformis by high-performance liquid chromatography. J Chroma A.911:55
Aboul-Enein HY, Serignese V (1996) Liquid chromatographic determination of taxol and related derivatives using a new polyfluorinated reversedpphase column. Analytica Chimica Acta. 319:187
Fu Y, Sun R, Zu Y (2009) Simultaneous determination of main taxoids in Taxus needles extracts by solid-phase extraction-high-performance liquid chromatography with pentafluorophenyl column. Biomed Chrom. 23:63–70
Shao L’Locke D (1997) Determination of paclitaxel and related taxanes in bulk drug and injectable dosage forms by reversed phase liquid chromatography. Anal Chem. 69:2008–2016
Badea I, Ciutaru D, Lazar L (2004) Rapid HPLC method for the determination of paclitaxel in pharmaceutical forms without separation. J Pharm Biomed Anal. 34:501–507
Ciutaru D, Badea I, Lazara L (2004) A HPLC validated assay of paclitaxel's related impurities in pharmaceutical forms containing Cremophor EL. J Pharm Biomed Anal. 34:493–499
United States Pharmacopoeia (2004) United States Pharmacopoeia Convention, pp 1394–1396
Sun P, Wang X, Alquier L (2008) Determination of relative response factors of impurities in paclitaxel with high performance liquid chromatography equipped with ultraviolet and charged aerosol detectors. J Chrom A. 1177:87–91
Beijing China (2010) Chinese Pharmacopoeia Commission. Beijing, Chinese Pharmacopoeia (Partα), pp 1007–1008
Volka K, Hill S, Kerns E (1997) Profiling degradants of paclitaxel using liquid chromatography–mass spectrometry and liquid chromatography–tandem mass spectrometry substructural techniques. J Chrom B Biomed Sci Appl. 696:99–115
Harmonised Tripartite Guideline ICH (2005) Validation of Analytical Procedures: Text and Methodology Q2(R1)
Acknowledgements
We are thankful for all those who have supported us during this research work.
Funding
There is no funding source for this project.
Author information
Authors and Affiliations
Contributions
NK conducted the literature study, designed, developed and validated the gas chromatographic method. NK and AR compiled, analyzed and interpreted the data. NK wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Noorbasha, K., Shaik, A. Determination of residual solvents in paclitaxel by headspace gas chromatography. Futur J Pharm Sci 7, 40 (2021). https://doi.org/10.1186/s43094-021-00186-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43094-021-00186-7