
Abstract
Background
A simple, rapid and accurate reverse phase RP-HPLC method for the validation of antitubercular drug bedaquiline fumarate (BQF) was developed. The stability and forced degradation behaviour of bedaquiline fumarate (BQF) in official dissolution media (0.01 N HCl) and methanol were assessed.
Results
The method provided linear responses within the concentration range which varies from 10 to 100 μg/mL with LOD values of 2.6 μg/mL and LOQ of 7.9 μg/mL. Mean percent recovery varied between 97.46 and 103.82% and precise mean RSD of 0.31% (repeatability) and 0.48% (intermediate precision). The method was validated for other parameters like specificity, system suitability and robustness. Potential degradation of the BQF when exposed to direct sunlight in official dissolution media was 90.75% and in methanolic stock solution was 91.98%. Acid-alkali degradation study showed 86.59% loss of BQF in 0.5 N NaOH, and in acidic environment (0.5 N HCl), there was 6.15% loss. Thermal degradation of BQF was 70.11% loss in official dissolution media at 80 °C. No significant change in drug concentrations was noted when exposed and protected from normal daylight at room temperature and at 2–8 °C temperature.
Conclusion
The developed method has significant applicability for the qualitative and quantitative determination of BQF in different pharmaceutical formulations in the official dissolution media in routine quality control analysis.
Similar content being viewed by others

Background
Mycobacterium tuberculosis (TB) is a highly notorious bacterium, popularly known as superbug which acts as causative agent for tuberculosis [1]. TB not only affects the lungs but also other parts of the body. One third of the population of the world is believed to be infected by TB with frequency of occurrence being 1% of the total population per annum [2]. About 1.3 million deaths were reported in 2016 out of 10 million cases of active TB. Greater than 95% deaths occurred in the developing countries whereas 50% in South Asian countries including India, China, Pakistan, Indonesia and the Philippines [3, 4].
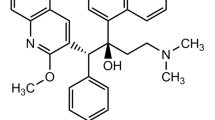
Bedaquiline fumarate (BQF) is a diarylquinoline derivative (Fig. 1) approved by the USFDA in 2012 with the accelerated approval pathway [5]. This drug was developed by Janssen Pharmaceutical (Johnson & Johnson) by Dr. Koen Andries and his team and marketed under the brand name ‘SIRTURO®’ for the treatment of multidrug resistance (MDR) pulmonary tuberculosis in combination with first line TB therapy [5, 6]. It is the first drug to be approved with a well-defined mechanism of action, for tuberculosis after 1998 (approval year of Rifapentine) [7]. Bedaquiline fumarate kills the superbug by inhibiting the enzyme ATP synthase which plays a vital role in the energy production. CYP3A4 enzyme of the cytochrome P450 family is responsible for the metabolism of BQF and generates M2 as the major metabolite with 6 times less activity [8]. Currently, BQF tablets are available with the strength of 100 mg, which needs to be taken 4 times in a day for the first 2 weeks of therapy followed by 200 mg, 3 times/week with a minimum gap of 48 h between subsequent doses for further duration of therapy. The drug should be prescribed under strict supervision with the special instruction to reserve BQF when an effective treatment by TB regimen cannot otherwise be provided [9, 10]. Bedaquiline belongs to Class-II of Biopharmaceutical Classification System (BCS), bearing poor aqueous solubility. Its bioavailability/dissolution rate is compromised (limited) when orally administered [11]. The official dissolution media for BQF is 0.01 N HCl (as per FDA), and methanol is commonly used as a solvent to make the stock solution during the development of analytical method. Thus, BQF has to be stable in official dissolution media and methanol during the study duration. The quantitative and qualitative analysis of BQF during dissolution study of marketed formulations as well as other novel formulations under development is necessary. There are, however, very few reported methods currently available in the literature. The methods currently available includes assay and content uniformity determination of BQF in triple combination spray-dried formulation for inhalation, effect of different chromatographic conditions on enantiomeric separation of BQF, separation BQF analogue diastereomer using polysaccharide based columns, and preclinical drug interaction assessment of BQF by LC-ESI-MS/MS bio-analytical method [12,13,14,15]. However, no method so far has been reported to evaluate the stability and forced degradation behaviour of BQF in official dissolution media and methanol.

Chemical structure of BQF
The current study was planned for the development and validation of a simple, accurate, precise and robust method to evaluate the stability and degradation behaviour of BQF in official media and methanol. As suggested above, few analytical methods are reported for BQF by RP-HPLC using PDA detector.
Methods
Materials and reagents
BQF was purchased from Clearsynth labs, Mumbai, India, for the study purpose. HPLC grade methanol and analytical grade acetic acid, hydrochloric acid (HCl) and sodium hydroxide (NaOH) were used in the study and purchased from Merck India Ltd. Ammonium acetate was obtained from Himedia, Mumbai, India. Triple distilled water was obtained from in house Milli-Q water purifier system (Merck, Prague, Czech Republic) which was used for the preparation of mobile phase and other solutions.
Instrumentation
The liquid chromatographic system used in the present work was consisted of Waters 1525 binary pump system with inline degasser, Waters 2707 auto sampler, column oven, Waters 2998 Photodiode Array (PDA) detector and the EMPOWER 2.0 software for data acquisition and processing (all Waters Corporation, Milford, MA, USA). The analysis was performed at 40 °C column oven temperature on a Sunfire C18 column (250 mm × 4.6 mm, 5 μm particle size). A Sunfire C18 analytical guard column (12.5 mm × 4.6 mm, 5 μm particle size) packed with the same sorbent was connected before the analytical column. The isocratic mode was employed for all the experiments.
Chromatographic conditions
The mobile phase consisted of 10 mM ammonium acetate buffer as aqueous phase (A) and methanol as organic phase (B). The aqueous phase was maintained at pH 4.5, adjusting with acetic acid and filtered through 0.45 μ membrane filter by applying vacuum. Both the phases were degassed prior to use by applying ultrasonication for 10 min. Isocratic mode was employed for the elution of analyte (BQF) in the ratio of 15:85 v/v (A:B), and flow rate was set at 1.2 ml/min. Elution was performed on Sunfire C18 column with dimensions of 250 × 4.5 mm and 5 μm particle size. The column oven temperature was set at 40 °C. The analyte detection was carried out using PDA detector with spectral range of 210 to 400 nm, and chromatogram was extracted at 226-nm wavelength. The injection volume was set to a constant volume of 10 μl for each injection.
Preparation of standard stock solution and calibration curve
Stock solution of 1 mg/mL was prepared by solubilising 6.1 mg of BQF (equivalent to 5.0 mg of free base) in 5 ml of HPLC methanol. The prepared stock was kept in a refrigerator at 2 to 8 °C till it is being used. The standard solutions were prepared by diluting the stock solution with methanol to reach concentration range 10–100 μg/mL for BQF. These standard samples were considered at eight different levels, i.e. 10%, 25%, 50%, 75%, 100%, 125%, 150% and 200% of the target concentration (50 μg/mL).
Method validation
The method validation protocol was set on the basis of recommendations provided by the USFDA and ICH guidelines.
System suitability
The verification of the resolution and reproducibility was evaluated by performing the system suitability test. The standard concentration of 10 μg/mL in methanol was injected in six replicates (n = 6). The RSD of retention time and the area was determined and compared with the ICH criteria for the system suitability.
Linearity
The linearity of the BQF was obtained by making the calibration standard curve in HPLC grade methanol. The calibration standards were prepared as mentioned previously in four replicates (n = 4), duplicate on day 1 and duplicate on day 2 in order to investigate the interday variability. The samples were injected to HPLC covering the range of 10 to 100 μg/mL. The peak area obtained from each standard was plotted against the concentration, and linearity equation along with regression coefficient (r2) was obtained. The linearity was evaluated by linear regression analysis.
Limit of detection (LOD) and limit of quantification (LOQ)
LOD and LOQ are denoting ability of the method to reliably detect and quantitate the smallest amount of analyte, respectively. The LOD and LOQ of the proposed method were calculated by using standard deviation of response and slope of regression line by using the following equations.
where σ is the standard deviation of response and s is the slope of regression line.
Accuracy
Accuracy of the developed analytical method was evaluated by injecting the analyte at three different levels of target concentration (80%, 100% and 120%) as per the ICH guideline. The samples at each level were prepared in six replicates (n = 6), and recovery was calculated from the obtained peak area using linearity equation.
Precision
The repeatability and intermediate precision were determined by considering the injection of analyte at three levels (low, medium and high, i.e. 10, 50 and 100 μg/mL, (n = 3)) on three different days. The concentrations were calculated from standard curve, and results were noted as relative standard deviation (RSD).
Specificity
The specificity of the method was established by injecting the blank sample without analyte, and the interference at the retention time (tR) of analyte was investigated. The blank methanol was injected in triplicate to evaluate whether there are any other interfering substances eluting at the tR of analyte.
Robustness
The robustness of the method was studied by deliberately changing in previously mentioned parameters. The standard concentration of 10 μg/mL was injected in triplicate, and the change in RSD was observed.
Mobile phase composition
The effect of changes in the mobile phase composition was evaluated by changing the composition by ± 3% from the actual composition (15:85% v/v of A:B).
Column temperature
The robustness of the method to the change in column temperature was evaluated to see its effect on the peak retention and peak area. The temperature was varied at points includes 1 °C above and 1 °C below fixed method temperature (40 °C). The variation in room temperature and the area was reported as RSD.
pH of mobile phase
The method sensitivity for change in pH of the mobile phase was evaluated to check the robustness of the developed method. The pH of the mobile phase was varied by ± 0.1 from the actual method pH (4.5).
Flow rate
The method sensitivity for change in flow rate was also evaluated. For this purpose, the flow rate was varied by 0.1 ml/min above and below the method actual flow rate (1.2 ml/min). The results are reported as the RSD in peak tR and peak area.
Accelerated degradation studies
All degradation experiments were performed in solution state at a drug concentration of 50 μg/mL, and injections were made to HPLC (10 × dilution). For acid-alkali degradation, 5 ml of stock solution of 50 μg/mL analyte in 0.5 N HCl and 0.5 N NaOH was prepared in three replicates and sonicated for 2 h followed by injection in HPLC (after dilution in mobile phase). For photo and thermal decomposition of analyte, 50 μg/mL stock solutions was prepared 0.01 N HCl (official dissolution media) and in methanol in triplicates. The stocks were exposed to direct sunlight for 12 h for photo stability and to 80 °C for thermal stability evaluation for 4 h, respectively. The percent degradation of BQF was calculated from the linear equation.
Stability study
The short-term stability study was performed to evaluate the stability of analyte in official dissolution media (0.01 N HCl) and in methanol up to 72 h at room temperature. The photo and thermal stability were also investigated in the same solvents by keeping the samples protected from normal daylight and keeping sample at refrigerator temperature (2 to 8 °C) respectively. For this, three stocks of 50 μg/ml were prepared in 0.01 N HCl and in methanol (normal daylight exposed, protected from daylight and freeze sample) directly from the 1 mg/ml methanolic stock solution (n = 3). The samples were withdrawn from each stock at a predetermined time period (1 h, 2 h, 4 h, 8 h, 24 h, 28 h and 72 h), diluted in HPLC grade methanol and injected in HPLC for quantification.
Results
Optimization of chromatographic condition
We developed an analytical method for the qualitative and quantitative evaluation of forced degradation behaviour and stability of BQF in official dissolution medium and methanol. The method validation was carried out as per the ICH guidelines and USFDA recommendations. The preliminary trials for the optimization of peak shapes were carried out by using methanol as organic phase and triple distilled water as aqueous phase on Sunfire C18 column (250 mm × 4.6 mm, 5 μ). We got the acceptable peak shape with the elution of BQF at room temperature (RT) after 12.5 min. However, this mobile phase composition was not able to sustain the change in pH on addition of small amount of acid or base which may lead to change in peak retention. In order to resolve this issue, we decided to use buffer system. The use of phosphate buffer system was deliberately avoided due to the precipitation at higher organic content [16, 17]; instead, an ammonium acetate buffer (10 mM) with pH adjusted to 4.5 by acetic acid was used. The flow rate and mobile phase composition optimization was carried out to get good peak shape with acceptable fronting and tailing. The elution was also tried at different flow rates and oven temperature. Eventually, the acceptable peak symmetry and resolution was obtained by setting the flow rate at 1.2 ml/min with A (ammonium acetate buffer):B (methanol) ratio of 15:85% v/v and column oven temperature of 40 °C. The drug peak was evaluated at two wavelengths, i.e. at 226 nm and 333 nm using PDA detector. The drug absorption at 333 nm was found to be less compared to at 226 nm which made us to choose 226 nm wavelength for more sensitive analysis.
Validation of the method
The purpose of method validation was to confirm the suitability of the present method to fulfil the objectives mentioned previously, as per the regulatory guidelines. The developed method was extensively validated for system suitability, linearity, accuracy, precision, limits of detection (LOD) and quantification (LOQ), specificity and robustness. The precision was expressed in terms of relative standard deviation with respect to the intra- and interday variability at specific concentration. The accuracy was expressed in terms of percent recovery injecting the different levels. The post validated method was employed for analysing the degradation and stability samples.
System suitability
System suitability is an important part in the process of chromatographic method validation to verify the ability of proposed method to generate peak of interest with good resolution and high reproducibility. The test ensures performance of HPLC system and procedure to produce acceptable quality data [18, 19]. Six replicates of samples (10 μg/mL) were prepared from fresh standard solution (100 μg/mL) in methanol for determining the system suitability. The chromatogram obtained is shown in Fig. 2. The analysis was performed by evaluating the parameters such as resolution (Rs), capacity factor (k’), USP tailing factor (T), number of theoretical plates (N) and RSD of peak area and retention time for six replicates. The RSD calculated for peak area and retention time of BQF were found to be 0.71 and 0.24 respectively which was less than 1% (meets acceptance limit) suggesting reproducibility of BQF peak with approximately same area and tR during each injection of fixed concentration (supplementary data S1-table 1). The k’ value was found to be 2.4 which was well within the acceptable limit of greater than 2 in order to achieve good separation [20].The resolution (Rs) between the BQF peak and first eluted peak (unretained peak (t0)) was found to be greater than 2 representing good separation among them. The T value and N values were found to be less than 2 and greater than 2000 respectively, thereby following the acceptance criteria of guidelines [19,20,21]. The system suitability results for BQF are compared with regulatory acceptance limits and are shown in Table 1. The results suggest that the developed method fulfils all the criteria related to system suitability.

HPLC chromatogram of BQF demonstrating system suitability
Linearity
The calibration curve representing linearity of developed method was obtained by plotting different standard concentrations of BQF against peak area. The standard concentrations were consisting of range from 20 to 200% of target concentration (50 μg/mL), i.e. 10 to 100 μg/mL. Two replicates were prepared for each concentration on day 1 and day 2, and linearity was determined (Supplementary data S2-table 2). The calibration plot of BQF concentration vs. peak area was found to be linear within the abovementioned concentration range by employing linear simple regression least square method (Supplementary data S2-figure 1) [22,23,24]. The equation of regression line was found to be y = 100,483x – 76,726 with correlation coefficient (r2) of 0.9991 (n = 4) which is well above the limit provided by guideline [25]. The RSD of the entire standard was found to vary within 0.58–4.5% which indicates the reproducibility of the results.
Limit of detection (LOD) and limit of quantification (LOQ)
Several strategies for the determination of LOD and LOQ have been described in ICH guideline Q2B which includes signal to noise ratio, visual evaluation and use of standard deviation of response and slope of standard plot [26]. In the present study, LOD and LOQ were determined by using standard deviation and slope of equation by employing the formulae 3.3σ/s and 10σ/s respectively, where σ is the standard deviation of response and s is the slope. The calculated values for LOD and LOQ were found to be 2.6 μg/mL and 7.9 μg/mL respectively.
Accuracy
The recovery experiment was performed to assess accuracy of the proposed method [27]. Standard concentrations were spiked at three levels of target concentration (80, 100 and 120%) in five replicates and analysed for drug content (Supplementary data S3-table 3). The calculated drug concentrations were compared with the nominal concentrations, and percent recovery was calculated. The results showed mean percent recovery of 103.82%, 102.59% and 97.46% for 80, 100 and 120% levels, respectively. The mean percent recovery at all the three levels of five replicates fall within the acceptance limit of 90–110% (Supplementary data S3-table 4) as per regulatory guidelines [28].
Precision
The precision of developed method was evaluated by evaluating parameters such as repeatability and intermediate precision. The three replicates were chosen for the three different levels (low, medium and high) covering the range of 20, 50 and 150% of the target concentration. Minimum nine determinations were obtained per day by injecting and analysing each freshly prepared concentration in triplicate at three different days in order to evaluate interday variability (Supplementary data S4). The mean RSD values for repeatability of all the three levels at each day was found to be 0.31 (average RSD of intraday variability on each day at three levels as given in Supplementary data S4A-table 5), and the RSD for intermediate precision was found to be 0.48 representing good precision of the developed method among different days as shown in Supplementary data S4B-table 6 (acceptable intra- and interday variability).
Specificity
The absence of interference at the retention of analyte of interest (BQF) was demonstrated by injecting the blank sample which consists of diluents or other excipients that were present in the samples [17]. The present method for BQF showed no interference at the retention of analyte (Fig. 3). Additionally, percent recovery in the accuracy samples was found to be good (102.12%) which represents the elution of analyte at its retention of 7.52 without any interfering substances. Similarly, no interference of the degradation products during the stress study such as heat, light, acid (0.5 N HCl) and alkali (0.5 N NaOH) at the tR of pure analyte was observed which represents ability of developed method to separate all the other potential components that may be produced during sample processing. The purity of the analyte peak can be determined by performing peak purity analysis where higher purity angle values over purity threshold can be seen for pure peaks. This method of peak purity analysis using PDA detector proved to be very useful in finding out any other co-eluting component.

HPLC chromatogram demonstrating specificity of the method
Robustness
United States Pharmacopeia (USP) has defined robustness as the ability of analytical method to remain unaffected when small but deliberate changes were made in method parameters to provide reliability of the method during normal analysis [28]. To assess the reliability of proposed method for BQF, various parameters were investigated which includes mobile phase composition, column temperature, pH of mobile phase and flow rate. The lower level (20%) of target concentration was chosen for the assessment to pick up the minute variation. The mean percent recovery of three replicates and shift in tR were monitored as a response to deliberate changes in method parameters (Supplementary data S5-table 7). The mobile phase composition variation was made within ± 3% of set method composition (15:85% v/v for A:B). The percent recovery of 97.61 and 100.64 with average tR of 8.3 minute (min) and 12.6 min were obtained at ratio of 12:88% v/v and 18:82% v/v (A:B) respectively. The column temperature was changed by 1 °C above and below the set temperature of 40 °C which gave mean percent recovery of 96.04 (tR of 7.4) and 97.04 (tR of 7.5) without any significant change in the original tR for BQF. However, change in tR of BQF was observed from 7.5 to 6.8 and 6.9 min when pH of the mobile phase changed by 0.1 above (pH 4.6) and below (pH 4.4) actual method pH of 4.5 with mean percent recovery of 104.34 and 102.07 respectively. The change in mobile phase flow rate by ± 0.1 ml/min led to a significant effect on tR for BQF, i.e. 10.6 and 9.0 min for flow rate of 1.1 and 1.3 ml/min with acceptable mean percent recovery of 104.09 and 90.19, respectively. Looking at the overall results of the study, it was noted that deliberate changes made in the proposed study parameters significantly affects only the tR of BQF except in case with the column temperature which shows good temperature tolerance. However, mean percent recovery was found to remain unaffected in all the conditions with slight variability within the acceptable limit. These results suggest that the proposed method is reliable for changes in the parameters within the abovementioned range and presents good robustness.
Accelerated degradation studies
The accelerated degradation study was performed to assess the stability of BQF under stressed conditions such as acid-alkali hydrolysis, thermal degradation and photo degradation by direct sunlight (Fig. 4 and supplementary data S6-table 8, 9 and 10). Extensive degradation of BQF was observed under alkaline condition (0.5 N NaOH) with total loss of 86.59%. However, it was stable under acidic environment (0.5 N HCl) with the loss of only 6.15%. This suggests the susceptibility of BQF molecule towards alkaline environment and thus must be prevented. These results are in line with Committee for Medicinal Products for Human Use (CHMP) assessment report for SIRTURO published by European Medicines Agency (EMA) on 19 December 2013. The study in EMA report revealed extreme instability of BQF under basic condition along with significant degradation in acidic and neutral condition [22]. The degradative profile of drug obtained on exposure to direct sunlight (12 h) showed contradictory results to EMA report, where report suggested slight sensitivity of BQF towards photolysis. Our study, however, revealed extensive degradation with loss of about 91.48% in methanolic solution and 90.75% in 0.01 N HCl solution. The HPLC chromatogram of sunlight-exposed methanolic solution of BQF had shown about 12 degradation peaks, whereas 5 degradation peaks were observed in case of 0.01 N HCl. These contradictory results might be due to the exposure of BQF to light in different physical states (solid sample of BQF in case with SIRTURO and BQF solution in our study). Another attributing factor might be the use of different light source in the study. To obtain thermal degradation profile of BQF in methanol and 0.01 N HCl, samples were exposed to 80 °C temperature for 1 h and physical appearance of both solutions was observed. The methanolic solution was found clear, whereas turbidity was observed in case of 0.01 N HCl solution which was subjected to filtration followed by injection in HPLC with sufficient dilution in mobile phase. The turbidity represents the potential degradation of BQF in 0.01 N HCl when exposed to higher temperature which was not observed with the samples kept at room temperature. This incidence might be due to temperature-assisted acceleration of acid catalytic degradation of ionisable functional groups in BQF molecule, and the turbidity represents involvement of water as a reactant in hydrolytic reaction [23, 24]. The quantitative data of percent loss for BQF after thermal exposure revealed insignificant degradation in methanolic solution (0.1% degradation). On the other hand, 70.11% degradation was observed in case of 0.01 N HCl. These results suggested that BQF is susceptible to potential hydrolytic degradation in official dissolution media when exposed to higher temperature conditions which must be kept in mind while performing dissolution and solubility experiments of BQF.

Accelerated degradation study representing BQF degradation behaviour in 0.5 N HCl (a), 0.5 N NaOH (b), 0.01 N HCl exposed to direct sunlight (c), methanol exposed to direct sunlight (d)
Stability study
The short-term and long-term solution stability of BQF in 0.01 N HCl and methanol was evaluated under different storage conditions (at RT and 2–8 °C) for specified duration of 0 to 72 h. The effect of normal daylight exposure on BQF stability at RT was also assessed. The experiments were performed in three replicates, and data are demonstrated in Fig. 5 and supplementary data S7. The BQF solutions which were exposed to normal daylight at RT had shown 95.62 and 98.44% stability, whereas those which were protected from normal daylight were 99.60 and 98.15% stable after 72 h in 0.01 N HCl and methanol respectively. The samples stored at 2–8 °C were found to get precipitated in 0.01 N HCl after 1 h due to temperature effect whereas the same was 99.61% stable in case of methanolic solution after 1 h without any precipitation during the overall study duration after 72 h and found to be 97.67% stable. The results suggest that BQF solutions, both in 0.01 N HCl and methanol, were stable for the specified period of time at RT and 2–8 °C even after exposure to light without any significant degradation (no degradation peaks observed).

BQF stability in 0.01 N HCl and methanol at RT and 2–8 °C temperature under normal daylight light exposure and light protection
Discussion
It has been concluded from the study that developed and validated RP-HPLC method is simple, accurate, precise and reliable. The experiment evaluating specificity revealed that there were no other interfering components eluting at the tR of BQF which was 7.5 min. All the validation parameters assessed by this method were within the acceptable limit provided by regulatory guidelines. Simplicity, sensitivity, robustness and the use of less expensive reagents in the mobile phase preparation made this method more acceptable, as very limited studies are available in literature regarding RP-HPLC methods for the analysis of BQF. The developed and validated method has been successfully used in the quantitative determination of BQF in the forced degradation study without any interference of degradation products which further proves its specificity. The stability study of BQF was also evaluated using proposed method which revealed that BQF was stable for specified time period in official dissolution media and in methanol which is most commonly used solvent during stock solution preparation.
Conclusion
This study suggests the future applicability of this method for qualitative and quantitative determination of BQF in different pharmaceutical formulations in the official dissolution media in routine quality control analysis.
Availability of data and materials
All data and material is available upon request.
Abbreviations
- BQF:
-
Bedaquiline fumarate
- HPLC:
-
High-performance liquid chromatography
- TB:
-
Mycobacterium tuberculosis
- LOD:
-
Limit of detection
- LOQ:
-
Limit of quantification
- tR:
-
Retention time
References
Nazir T, Abraham S, Islam A (2012) Emergence of potential superbug Mycobacterium tuberculosis, lessons from New Delhi mutant-1 bacterial strains. Int J Health Sci (Qassim) 6(1):87–94. https://doi.org/10.12816/0005977
Lee SH (2016) Tuberculosis infection and latent tuberculosis. Tuberc Respir Dis (Seoul). 79(4):201–206. https://doi.org/10.4046/trd.2016.79.4.201
Jones JG, King TC (2015) Latent mycobacterium tuberculosis infection. N Engl J Med. 373(12):1178. https://doi.org/10.1056/NEJMc1508223
Raviglione MC, Kochi A, Snider DE (1995) Global epidemiology of tuberculosis: morbidity and mortality of a worldwide epidemic. JAMA. 273(3):220–226. https://doi.org/10.1001/jama.1995.03520270054031
Center of Disease Control (2013) Provisional CDC guidelines for the use and safety monitoring of bedaquiline fumarate (Sirturo) for the treatment of multidrug-resistant tuberculosis. MMWR Recomm Rep 62(RR-09):1–12
Jenkins HE, Tolman AW, Yuen CM, Parr JB, Keshavjee S, Pérez-Vélez CM, Pagano M, Becerra MC, Cohen T (2014) Incidence of multidrug-resistant tuberculosis disease in children: systematic review and global estimates. Lancet. 383(9928):1572–1579. https://doi.org/10.1016/S0140-6736(14)60195-1
Leibert E, Danckers M, Rom WN (2014) New drugs to treat multidrug-resistant tuberculosis: the case for bedaquiline. Ther Clin Risk Manag. 10:597–602. https://doi.org/10.2147/TCRM.S37743
Yadav S, Rawal G, Baxi M (2016) Bedaquiline: a novel antitubercular agent for the treatment of multidrug-resistant tuberculosis. J Clin Diagnostic Res. J Clin Diagn Res. 10(8):FM01–FM02. https://doi.org/10.7860/JCDR/2016/19052.8286
Svensson EM, Aweeka F, Park JG, Marzan F, Dooley KE, Karlsson MO (2013) Model-based estimates of the effects of efavirenz on bedaquiline pharmacokinetics and suggested dose adjustments for patients coinfected with HIV and tuberculosis. Antimicrob Agents Chemother. 57(6):2780–2787. https://doi.org/10.1128/AAC.00191-13
Fiorillo M, Lamb R, Tanowitz HB, Cappello AR, Martinez-Outschoorn UE, Sotgia F, Lisanti MP (2016). Bedaquiline, an FDA-approved antibiotic, inhibits mitochondrial function and potently blocks the proliferative expansion of stem-like cancer cells (CSCs). Aging (Albany NY). Aging (Albany NY). 8(8):1593-607. doi: 10.18632/aging.100983.
Xia X, Pethe K, Kim R, Ballell L, Barros D, Cechetto J, HeeKyoung Jeon, Kideok Kim, and Alfonso E, Garcia-Bennett (2014). Encapsulation of anti-tuberculosis drugs within mesoporous silica and intracellular antibacterial activities. Nanomaterials. Nanomaterials (Basel). 4(3): 813–826. doi: https://doi.org/10.3390/nano4030813
Douša M, Reitmajer J, Lustig P, Štefko M (2016) Effect of chromatographic conditions on enantioseparation of bedaquiline using polysaccharide-based chiral stationary phases in RP-HPLC. J Chromatogr Sci. 54(9):1501–1507. https://doi.org/10.1093/chromsci/bmw050
Momin MAM, Rangnekar B, Das SC (2018). Development and validation of a RP-HPLC method for simultaneous quantification of bedaquiline (TMC207), moxifloxacin and pyrazinamide in a pharmaceutical powder formulation for inhalation. J Liq Chromatogr Relat Technol. 41 (8), 415-421.doi.org/10.1080/10826076.2018.1437748
Jiang S, Wu C, Yu J, Sun T, Guo X (2019) Separation of eight bedaquiline analogue diastereomers by HPLC on an immobilized polysaccharide-based chiral stationary phase. Chirality. 31(1):72–78. https://doi.org/10.1002/chir.23037
Kotwal P, Magotra A, Dogra A, Sharma S, Gour A, Bhatt S, Wazir P, Singh PP, Singh G, Nandi U (2019) Assessment of preclinical drug interactions of bedaquiline by a highly sensitive LC-ESI-MS/MS based bioanalytical method. J Chromatogr B Analyt Technol Biomed Life Sci. 1112:48–55. https://doi.org/10.1016/j.jchromb.2019.02.022
Schellinger AP, Carr PW (2004) Solubility of buffers in aqueous-organic eluents for reversed-phase liquid chromatography. LCGC North America. 22(6):544–548
Ermer J, John JH (2006). Method validation in pharmaceutical analysis: a guide to best practice. Method Validation in Pharmaceutical Analysis: A Guide to Best Practice. ISBN: 978-3-527-60447-0, 418 Pages.
Wiggins DE (1991) System suitability in an optimized hplc system. J Liq Chromatogr 14:3045–3060. https://doi.org/10.1080/01483919108049375
Shabir GA (2003) Validation of high-performance liquid chromatography methods for pharmaceutical analysis: understanding the differences and similarities between validation requirements of the US Food and Drug Administration, the US Pharmacopeia and the International Conf. In: Journal of Chromatography A. J Chromatogr A. 987(1-2):57–66. https://doi.org/10.1016/s0021-9673(02)01536-4
Snyder LR, Glajch JL, Kirkland JJ (1997) Practical HPLC method Second Edition. Wiley-Interscience. https://doi.org/10.1002/9781118592014
Araujo P (2009) Key aspects of analytical method validation and linearity evaluation. J Chromatogr B Anal Technol Biomed Life Sci. 877(23):2224–2234. https://doi.org/10.1016/j.jchromb.2008.09.030
Schlossberg D, Samuel R (2017). SIRTURO (Bedaquiline). In: Antibiotics Manual. ISBN: 978-1-119-22075-6, page 480.
Blessy M, Patel RD, Prajapati PN, Agrawal YK (2014) Development of forced degradation and stability indicating studies of drugs - a review. Journal of Pharmaceutical Analysis. 4(3):159–165
Aubry AF., Tattersall P., Ruan J. (2009) Development of stability indicating methods. In: Huynh-Ba K. (eds) Handbook of stability testing in pharmaceutical development. Springer, New York, NY doi.org/10.1007/978-0-387-85627-8_7.
ICH (2015). ICH harmanized tripartite guideline validation of analytical procedures: Federal Register, Vol. 62, No. 96.
ICH Expert Working Group (1996). International Conference on Harmonization (ICH) of Technical Requirements for the Registration of Pharmaceuticals for Human Use, Validation of analytical procedures: text and methodology. ICH-Q2B. https://database.ich.org/sites/default/files/Q2_R1__Guideline.pdf
ICH (2005). ICH Topic Q2 (R1) Validation of analytical procedures : text and methodology. Int Conf Harmon. https://www.gmp-compliance. org /guidemgr /files /Q2(R1). pdf
Shabir GA (2004) Step-by-step analytical methods validation and protocol in the quality system compliance industry. J Validation Tech. 10:314–324
Acknowledgements
None
Funding
VP and GP were awarded fellowship by Department of Pharmaceutical, Ministry of Chemical and Fertilizers, Government of India. The instrument, chemicals, and operational costs were borne National Institute of Pharmaceutical Education and Research, Raebareli, India.
Author information
Authors and Affiliations
Contributions
SJSF conceptualized the project and gave technical inputs in conducting the study and preparing manuscript. VP performed the study and prepared manuscript. GP gave technical inputs in conducting the study. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Supplementary information.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pardhi, V., Pant, G. & Flora, S.J.S. RP-HPLC method development and validation for bedaquiline fumarate to evaluate its forced degradation behaviour and stability in official dissolution media. Futur J Pharm Sci 6, 42 (2020). https://doi.org/10.1186/s43094-020-00061-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43094-020-00061-x