
Abstract
Background
COVID-19 was declared a pandemic by the World Health Organisation in 2020 after its outbreak in December 2019 in Wuhan, China. Since researchers have been working to develop specific drugs to cure COVID-19. COVID-19 is caused by the severe acute respiratory cornonavirus2 or popularly known as SARS-CoV2 attacking the ACE2 receptor in the human respiratory system. The main protease translated by the viral genome is a highly conserved protein that plays a crucial role in viral protein replication and transcription. Compounds such as Darunavir and danoprevir have been tested to show potential biological activity against the viral protein, but a high mutation rate defies a permanent solution to this problem.
Results
In this study, virtual screening of natural ligands (around 170,000 molecules) and FDA-approved repurposed drugs retrieved from ZINC Database was carried out against SARS-CoV2 main protease (PDB ID: 7DJR). Molecular coupling was performed for the top three ligands, where ZINC70699832 showed a significantly good binding affinity of − 11.05 kcal/mol. It has shown an interaction affinity for the residues THR-25, PHE-140, LEU-141, ASN-142, GLY-143, SER-144, CYS-145, MET-165, GLU-166, GLN-189 and GLN-192. The molecular dynamic simulation was also performed using GROMACS, for all complexes where the ZINC70699832–7DJR complex showed stability in terms of root mean square deviation.
Conclusion
The study recommends that ZINC70699832 has great potential to serve as a potent inhibitor of the main protease of SARS-CoV2 main protease.
Similar content being viewed by others

1 Background
Coronavirus Disease or COVID-19 had its first reported cases in Wuhan, China, in December 2019. It spreads like a forest fire and was soon declared a global pandemic by the World Health Organization (WHO). A virus causes this disease, severe acute respiratory syndrome coronavirus 2 (SARS-CoV2). SARS-CoV2 is a single-stranded, wrapped, unbroken, positive sense RNA virus with about 30,000 nucleotides coding for 9860 amino acids [1]. The appearance of the virus is somewhat like a crown due to the presence of the ‘spike’ glycoprotein. The virus genome codes for several structural proteins like the spike protein, membrane proteins, envelope proteins, nucleocapsid proteins, and accessory proteins. The spike (S) glycoprotein plays an important role in virus infectivity, as one of its prime functions is to be the prime corrupter of host immunity and the identification of target receptor [2]. It is observed that for successful entry into cell targets of human host the S-Glycoprotein and the host trans-membrane serine protease 2 (TMPRSS2) play a crucial role in binding to the angiotensin-converting enzyme 2 (ACE2) receptors [3,4,5]. The SARS-CoV2 genome has 10 open reading frames (ORF). ORF1a/b consists of about 2/3rd of viral RNA that codes for polyprotein 1a, polyprotein 1b and 1–16 non-structural proteins. The remaining ORFs code for structural proteins, such as spike proteins, membrane proteins, envelope proteins, nucleocapsid proteins, and accessory proteins. ORF1ab is translated into the pp1ab polyprotein through the 1-ribosomal frame shift mechanism [6,7,8] followed by proteolytic processing that results in the formation of the main protease Mpro, also called 3C-like protease(3CLpro). Mpro is responsible for proteolytic processing [9,10,11,12] (cleaving) at 11 sites that take part in the formation of replicase transcriptase complex. These complexes are vital for virus replication. Mpro is found to be an essential expression of the viral genome replication and is coded by the nsp5 gene in the viral genome [9]. The Mpro protease has a mass of around 33.8 kDa [10] and is distinguished as a self-cleavage protein [11, 12]. It consists of a homodimer subdivided into two protomers (A and B) that have 3 well-defined domains (Additional file 1: Fig. S1) [13]. The first two domains I and II are made of β-barrels forming a chymotrypsin structure and bearing a catalytic couple histidine 41 (HIS41), and cysteine 145 (CYS145) [6,7,8,9,10,11,12,13,14]. Domain III is made up of α-helices [15]. For catalytic interaction, Mpro needs to dimerize, establishing interactions with both N- and C- terminal domains of the other protomer [16].
It is well known that the main protease (Mpro) of SARS-CoV-2 plays a crucial role in the maturation of several viral proteins like such as RNA-dependent RNA polymerase (RdRp), and Nsp4-Nsp16. The dependency of the virus on Mpro, and given that no human proteases share similarity with it, makes this protein an optimistic drug target [17,18,19], and it is highly preserved (96.1% similarity) among coronaviruses [20]. Hence, there have been efforts to discover therapeutic candidates targeting Mpro using various computer aided drug designing methods like virtual screening methods based on pharmacophore, molecular docking, and molecular dynamic simulation [21,22,23,24,25,26,27,28]. Drugs such as Danoprevir, which is legally used for the treatment of chronic Hepatitis C in China, and Darunavir, which inhibits the maturation of virus particles by obstructing polypeptide cleavage in infected cells [29], have been used as repurposed drugs to inhibit virus protease clinically [30, 31]. Su et al. [32] have repurposed the inhibitory potential of Shuanghuanglian oral preparation a traditional Chinese patented medicine for 3CL protease, where Baicalein an active compound from the extract was observed productively embedded in the core of the substrate-binding pocket by interacting with two catalytic residues acting as a ‘blockade’ in front of the catalytic dyad, preventing substrate access within the active site. Similarly, Al-Zahrani [33] obtained 51 phytochemicals from the extract of Juniperus procera and docked them against the main protease of COVID-19. Among them, rutin (Additional file 1: Fig. S2) and lopinavir emerged as the best performing molecules.
Rutin, or rutoside or sophorin, is a glycoside that combines the flavonol quercetin and the disaccharide rutinose. Rutin and many other flavonols are under initial clinical research for their potential biological activity in post-thrombotic syndrome, venous insufficiency, and endothelial dysfunction. However, there was no prime evidence for safe and effective use as of 2018 [34,35,36]. Compared to other flavonols, rutin has a lower bioavailability due to poor absorption, high metabolism, and rapid excretion, making it unsuitable for therapeutic use [34]. Although many in silico studies have been conducted for the therapeutic applications of Rutin for the main protease SARS-CoV-2 [37,38,39,40,41], there is an acute need to conduct more experiments towards bioavailability.
2 Methods
The graphical representation of the materials and method can be expressed as in Fig. 1.

Working pipeline for proteomic investigation
2.1 Protein selection and structure preparation
Natural compounds were analysed using in silico approaches using the crystal structure of SARS-CoV-2 main protease (no ligand) (PDB ID–7DJR) [42] involved in viral replication of the SARS-CoV-2 genome that were downloaded in PDB format from the RCSB Protein Data Bank website (https://www.rcsb.org/) R-value work was 0.166. R-value free was 0.201 with a resolution of 1.45 Å selected for the present study. Specifically, this protein was chosen because not enough in silico studies have been performed using this protein and sole intention of the study is to inhibit the disease in its premature state, i.e. the monomer state. 7DJR has only one chain used to prepare macromolecules, and other coexisting water molecules and non-standard residues were removed using Biovia Discovery Studio 2021 Client. To produce a protonated state at physiological pH, build up geometry optimisation, addition of polar hydrogen, Kollman charges and Gasteiger charges, one uses Autodock4.2.
The three-dimensional structures of all the Natural Products and FDA-approved [43, 44] were retrieved from the ZINC database (https://zinc.docking.org) [45]. Energy minimisation, geometrical conformation, and hydrogen bond were made and the file format was converted from SDF to PDBQT using the Open Babel programme [46]. In the complete study, rutin (ZINC4096846) along with the FDA-approved drugs was considered as reference molecules.
2.2 Virtual screening
In order to investigate biological activity, the prime objective of molecular docking was to assess the binding interaction between the protein and ligand molecules. For the primary screening of the library, AutoDock Vina [47, 48] (an open-source programme for molecular docking) was used. Due to the massive size of the library, the process had to be automated. For automation purposes, a shell script was written that iterates the process of screening and stores results in a different directory.
2.3 Molecular docking
AutoDock4 [49] was used to perform individual molecular docking of top-screened ligand molecules (that is, natural products and FDA drugs) to gain confidence in the results. All the molecular docking was performed using a Genetic Algorithm, and exhaustiveness (or no. of runs) was set to 100. The size of the grid was adjusted according to the receptor binding pocket at coordinate X, Y, Z that were set around the centroid of the active site to centre X = 10.430, Y = − 0.021, Z = 20.536 and dimension coordinates at X = 72, Y = 64, Z = 60 (as in Additional file 1: Fig. S4). The complexes of all the files were obtained and were visualised with the help of PyMOL.
2.4 Molecular dynamic simulation
Molecular dynamics simulation studies were carried out by using GROMACS [50]. Swiss Param (https://www.swissparam.ch/) [51] server was used for ligand topology generation. The MOL2 coordinates of the ligand molecule were uploaded and the server provided the zip file for the ligand topology. The force field was set to CHARMM27 [52], the water model was TIP3, the box type was set to be cubic for the apo-protein and the dodecahedron for the complexes, respectively, the salt type was NaCl, the energy minimisation steps were set to be 50,000, the equilibration of NPT and NVT was carried out at 300 K with a simulation time of 100 ns. This process was repeated for all of the complexes, including the reference molecule, i.e. Rutin, FDA drug, and the apo-protein as well. All the results were then individually analysed.
2.5 Physiological parameters
Pharmacokinetic studies and toxicological characteristics are an important criterion for the selection of potential drug candidates. As an alternative to clinical trials, computational methods were developed to assess the bioactivity of a potential drug candidate [53]. ADMETlab 2.0 (https://admetmesh.scbdd.com/) [54] is an online ADME predictor that takes smile notation as input and tabulates the result of physiological characters.
3 Results
3.1 Virtual screening and molecular docking
The results observed for virtual screening were recorded considering the exhaustiveness to 8, and the substantial data set was tabulated, and the ZINC85893430, ZINC70699832 and ZINC70669786 (structures given below Fig. 2) were recorded as top 3 molecules with − 12.8, − 11.7 and − 11.5 kcal/mol ΔG◦, respectively. These three top molecules were utilised for further experiments against SARS-CoV-2 Mpro. Whereas rutin and danoprevir (Additional file 1: Table S1) had significantly less the binding affinity of − 7.39 kcal/mol ΔG0 and − 9.8 kcal/mol ΔG0.

Top three screened ligands (A ZINC70669786, B ZINC70699832, C ZINC85893430)
To increase the confidence in the observed binding affinities, another docking with an iteration of 100 Genetic Algorithms run was performed and the results obtained for the above molecules were tabulated (Table 1).
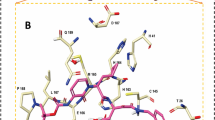
The complexes received were uploaded to the protein–ligand interaction profiler (PLIP) [55], an online web server to study the protein–ligand interaction. The output “. pse” file was visualised using PyMOL (Fig. 3),

Complex formed by A ZINC70699832, B ZINC70669786, C ZINC85893430, D ZINC4096846 (Rutin), E Danoprevir (FDA drug) visualised in PyMOL
3.2 Molecular dynamic simulation
The molecular dynamic simulation of all the samples was carried out on GROMACS using the CHARM27 forcefield, water model was TIP3, the box-type was Cubic and Dodecahedron, the salt-type was NaCl, the energy minimisation steps were set to be 5000, NPT and NVT equilibration was carried out at 300 K with a simulation time of 100 ns. The results obtained are shown below (Figs. 4, 5, and 6).

RMSD comparison of protein–ligand complexes with the apoprotein. A ZINC70699832, B ZINC70669786

RMSD Comparison of protein–ligand complexes with the apo-protein. A ZINC85893430, B Danoprevir (highest screened FDA drug)

RMSD Comparison of protein–ligand complexes with the apo-protein. A ZINC4096846 (Rutin)
3.3 Physiological parameters
The physiological quality or ADMET scores of the ZINC molecules were examined and tabulated from ADMETlab 2.0 on the basis of molecular weight, Log P, and Lipinski drug likeliness. From Table 2, the data suggest that among all the compounds ZINC70699832 has the highest potency to be a drug molecule due to least violations.
4 Discussion
4.1 Molecular docking
It is observed that among complexes (Fig. 3) ZINC70669786 has the highest binding energy of − 12.03 kcal/mol but due to its large size and sterically strained structure it is not able to fit itself in the active cavity and thus, has very few hydrogen bonds. Therefore, ZINC70669786 cannot interact with the crucial residues such as GLU-166 responsible for dimerisation of the protein [56], GLN-189 which is responsible for catalysis [57], HIS-41 and CYS-145 responsible for the formation of the catalytic dyad at the N and C terminal of the protein [6,7,8,9,10,11,12,13,14]. On the other hand, ZINC70699832 has shown a binding affinity of − 11.05 kcal/mol and interactions at all the critical active site residues, while ZINC4096846 (Rutin, reference) has also interacted with critical residues but failed to interact with GLN-189 responsible for catalysis, unlike ZINC70669832 and also has a very low binding affinity of − 7.39 kcal/mol. Both of the molecules have a perfect fitting in the active site cavity. In addition to conventional hydrogen bonds and hydrophobic interactions reported in the active site cavity, a π-stacking and a salt bridge formation over HIS-41 residue can also be seen. π-stacking is a non-bonding interaction, whereas a salt bridge is an ionic interaction both seemingly increase the stability of the complexes formed. ZINC85893430 and the FDA-approved drug danoprevir have a binding affinity of − 10.91 kcal/mol and − 9.8 kcal/mol, respectively, but due to their large size and sterically strained structure, ZINC85893430 and danoprevir are unable to fit themselves in the active site cavity. It attaches itself to a completely different position on the protein that may have some unknown function.
4.2 Molecular dynamic simulation
Figure 4 depicts the RMSD comparison of complexes formed by ZINC70699832 and ZINC70669786 having a stable deviation under 2 Å and 4 Å, respectively. The spikes in the graphs can be explained by the high presence of loops in the protein structure. Complex like ZINC85893430 due to their steric properties and massive size was not able to pull off a stable simulation and has shown considerable high deviation like 40 Å as shown in Fig. 5A. Control setups like danoprevir have shown a stable RMSD graph in Fig. 5B, but the stability difference is much higher (around 10 Å) from that of the apoprotein molecule, thus deeming it unfit to be a repurposed drug. The other control molecule ZINC4096846 or rutin has a seemingly unstable simulation pattern as shown in Fig. 6A. The RMSD differences between the apoprotein and protein-rutin complexes are not very high, but the complex struggled to attain stability throughout the simulation. From the above representation of RMSD graphs, judging an explicit drug molecule might be a difficult task but among the complexes, and it is evident that ZINC70699832 has shown better stability than the rest of the samples as well as control molecules.
5 Conclusion
This study mainly focuses on a swift pipeline for drug discovery. The proposed molecule ZINC70699832 has an optimum binding affinity of − 11.05 kcal/mol, covering almost all the important residues in the active site cavity of the protein like GLN-189, GLU-166, HIS-41, CYS-145 just like the reference molecule. The protein-ZINC70699832 complex has also shown a stable molecular dynamic simulation rather than a protein-reference complex. Drug likeliness of this compound is also higher than the others. This molecule can be used as a lead in a pharmacophoric study to produce even better results than ZINC70699832.
Availability of data and materials
Availability of data that supports the findings of this study is available from the corresponding author, upon reasonable request.
Abbreviations
- COVID:
-
Corona Virus Disease
- WHO:
-
World Health Organisation
- SARS-CoV2:
-
Severe acute respiratory syndrome Coronavirus 2
- ACE2:
-
Angiotensin-converting Enzyme2
- Mpro :
-
Main protease
- MD:
-
Molecular dynamics
- RMSD:
-
Root mean square deviation
- RMSF:
-
Root mean square fluctuation
- RNA:
-
Ribonucleic acid
- TMPRSS2:
-
Transmembrane protease serine 2
- ORF:
-
Open reading frame
- RdRp:
-
RNA-dependent RNA polymerase
- ADMET:
-
Absorption, Distribution, Metabolism, Excretion, Toxicity
- PDB:
-
Protein Data Bank
- RCBS:
-
Research Collaboratory for Structural Bioinformatics
- SPC:
-
Simple point charge
- NVT:
-
Number, constant volume and constant temperature
- NPT:
-
Number, constant pressure and constant temperature
- MOW:
-
Molecular weight
- FDA:
-
Food Drug Administration
- THR:
-
Threonine
- PHE:
-
Phenylalanine
- LEU:
-
Leucine
- ASN:
-
Asparagine
- GLY:
-
Glycine
- SER:
-
Serine
- CYS:
-
Cysteine
- MET:
-
Methionine
- GLU:
-
Glutamic acid
- GLN:
-
Glutamine
- ARG:
-
Arginine
- HIS:
-
Histidine
- LYS:
-
Lysine
References
Chan JFW, Kok KH, Zhu Z, Chu H, To KKW, Yuan S, Yuen KY (2020) Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microb Infect 9(1):221–236. https://doi.org/10.1080/22221751.2020.1719902
Agrawal PK, Agrawal C, Blunden G (2021) Rutin: a potential antiviral for repurposing as a SARS-CoV-2 main protease (Mpro) inhibitor. Nat Prod Commun 16(4):1934578X21991723. https://doi.org/10.1177/1934578X21991723
Du L, He Y, Zhou Y, Liu S, Zheng BJ, Jiang S (2009) The spike protein of SARS-CoV—a target for vaccine and therapeutic development. Nat Rev Microbiol 7(3):226–236. https://doi.org/10.1038/nrmicro2090
Wrapp D et al (2020) Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367(6483):1260–1263. https://doi.org/10.1126/science.abb2507
Hoffmann M et al (2020) SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181(2):271–280. https://doi.org/10.1016/j.cell.2020.02.052
Zhou P et al (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579(7798):270–273. https://doi.org/10.1038/s41586-020-2012-7
Wu F et al (2020) Author Correction: a new coronavirus associated with human respiratory disease in China. Nature 580(7803):E7. https://doi.org/10.1038/s41586-020-2008-3
Pillaiyar T, Manickam M, Namasivayam V, Hayashi Y, Jung SH (2016) An overview of severe acute respiratory syndrome–coronavirus (SARS-CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J Med Chem 59(14):6595–6628. https://doi.org/10.1021/acs.jmedchem.5b01461
Bzówka M, Mitusinska K, Raczynska A, Samol A, Tuszynski J, Gora A (2020) Molecular dynamics simulations indicate the SARS-CoV-2 Mpro is not a viable target for small-molecule inhibitors design. BioRxiv. https://doi.org/10.3390/ijms21093099
Jin Z et al (2020) Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 582(7811):289–293. https://doi.org/10.1038/s41586-020-2223-y
Meng T et al (2020) The insert sequence in SARS-CoV-2 enhances spike protein cleavage by TMPRSS. biorxiv, 2020-02. https://doi.org/10.1101/2020.02.08.926006
Kang S et al (2020) Crystal structure of SARS-CoV-2 nucleocapsid protein RNA binding domain reveals potential unique drug targeting sites. Acta Pharm Sin B 10(7):1228–1238. https://doi.org/10.1016/j.apsb.2020.04.009
Ionescu MI (2020) An overview of the crystallized structures of the SARS-CoV-2. Protein J 39(6):600–618. https://doi.org/10.1007/s10930-020-09933-w
Augustin TL, Hajbabaie R, Harper MT, Rahman T (2020) Novel small-molecule scaffolds as candidates against the sars coronavirus 2 main protease: a fragment-guided in silico approach. Molecules 25(23):5501. https://doi.org/10.3390/molecules25235501
Zhong N et al (2008) Without its N-finger, the main protease of severe acute respiratory syndrome coronavirus can form a novel dimer through its C-terminal domain. J Virol 82(9):4227–4234. https://doi.org/10.1128/JVI.02612-07
Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R (2003) Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science 300(5626):1763–1767. https://doi.org/10.1126/science.1085658
Skorenski M, Sienczyk M (2013) Viral proteases as targets for drug design. Curr Pharm Des 19(6):1126–1153. https://doi.org/10.2174/1381612811319060013
Zhang P et al (2020) Evaluation of recombinant nucleocapsid and spike proteins for serological diagnosis of novel coronavirus disease 2019 (COVID-19). MedRxiv, 2020-03. https://doi.org/10.1101/2020.03.17.20036954
Xu X et al (2020) Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci China Life Sci 63:457–460. https://doi.org/10.1007/s11427-020-1637-5
Xue X et al (2008) Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. J Virol 82(5):2515–2527. https://doi.org/10.1128/JVI.02114-07
Zhang L et al (2020) Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 368(6489):409–412. https://doi.org/10.1126/science.abb3405
Fauquet CM, Fargette D (2005) International Committee on taxonomy of viruses and the 3142 unassigned species. Virol J 2:1–10. https://doi.org/10.1186/1743-422X-2-64
Nukoolkarn V, Lee VS, Malaisree M, Aruksakulwong O, Hannongbua S (2008) Molecular dynamic simulations analysis of ritronavir and lopinavir as SARS-CoV 3CLpro inhibitors. J Theor Biol 254(4):861–867. https://doi.org/10.1016/j.jtbi.2008.07.030
Mirza MU, Froeyen M (2020) Structural elucidation of SARS-CoV-2 vital proteins: computational methods reveal potential drug candidates against main protease, Nsp12 polymerase and Nsp13 helicase. J Pharm Anal 10(4):320–328. https://doi.org/10.1016/j.jpha.2020.04.008
Kong R et al (2020) COVID-19 docking server: a meta server for docking small molecules, peptides and antibodies against potential targets of COVID-19. Bioinformatics 36(20):5109–5111. https://doi.org/10.1093/bioinformatics/btaa645
Gimeno A et al (2020) Prediction of novel inhibitors of the main protease (M-pro) of SARS-CoV-2 through consensus docking and drug reposition. Int J Mol Sci 21(11):3793. https://doi.org/10.3390/ijms21113793
Singh E et al (2020) A comprehensive review on promising anti-viral therapeutic candidates identified against main protease from SARS-CoV-2 through various computational methods. J Genet Eng Biotechnol 18(1):1–12. https://doi.org/10.1186/s43141-020-00085-z
Li Z, Li X, Huang YY, Wu Y, Liu R, Zhou L, Luo HB (2020) Identify potent SARS-CoV-2 main protease inhibitors via accelerated free energy perturbation-based virtual screening of existing drugs. Proc Natl Acad Sci 117(44):27381–27387. https://doi.org/10.1073/pnas.2010470117
Teoh SL, Lim YH, Lai NM, Lee SW (2020) Directly acting antivirals for COVID-19: where do we stand? Front Microbiol 11:1857. https://doi.org/10.3389/fmicb.2020.01857
Wang S, Guo L, Chen L, Liu W, Cao Y, Zhang J, Feng L (2020) A case report of neonatal COVID-19 infection in China. Clin Infect Dis 71(15):853–857. https://doi.org/10.1093/cid/ciaa225
Cao B, Wang Y, Wen D, Liu W, Wang J, Fan G, Wang C (2020) A trial of lopinavir–ritonavir in adults hospitalized with severe Covid-19. N Engl J Med. https://doi.org/10.1056/NEJMoa2001282
Su HX, Yao S, Zhao WF, Li MJ, Liu J, Shang WJ, Xu YC (2020) Anti-SARS-CoV-2 activities in vitro of Shuanghuanglian preparations and bioactive ingredients. Acta Pharmacol Sin 41(9):1167–1177. https://doi.org/10.1038/s41401-020-0483-6
Al-Zahrani AA (2020) Rutin as a promising inhibitor of main protease and other protein targets of Covid-19: in silico study. Nat Prod Commun 15(9):1934578X20953951. https://doi.org/10.1177/1934578X20953951
Vitamin A (2021) Micronutrient Information Center, Linus Pauling Institute, Oregon State University, Corvallis. 2015. Archived from the original on April 27
Morling JR, Broderick C, Yeoh SE, Kolbach DN (2018) Rutosides for treatment of post-thrombotic syndrome. Cochrane Database Syst Rev. https://doi.org/10.1002/14651858.CD005625.pub4
Martinez-Zapata MJ, Cosp XB, Moreno RM, Vargas E, Capellà D (2005) Phlebotonics for venous insufficiency. Cochrane Database Syst Rev. https://doi.org/10.1002/14651858.CD003229.pub3
Cherrak SA, Merzouk H, Mokhtari-Soulimane N (2020) Potential bioactive glycosylated flavonoids as SARS-CoV-2 main protease inhibitors: a molecular docking and simulation studies. PLoS ONE 15(10):e0240653. https://doi.org/10.1371/journal.pone.0240653
Xu Z, Yang L, Zhang X, Zhang Q, Yang Z, Liu Y, Liu W (2020) Discovery of potential flavonoid inhibitors against COVID-19 3CL proteinase based on virtual screening strategy. Front Mol Biosci 7:556481. https://doi.org/10.3389/fmolb.2020.556481
Hu X, Cai X, Song X, Li C, Zhao J, Luo W, He Z (2020) Possible SARS-coronavirus 2 inhibitor revealed by simulated molecular docking to viral main protease and host toll-like receptor. Futur Virol 15(6):359–368. https://doi.org/10.2217/fvl-2020-0099
Huynh T, Wang H, Luan B (2020) Structure-based lead optimization of herbal medicine rutin for inhibiting SARS-CoV-2’s main protease. Phys Chem Chem Phys 22(43):25335–25343. https://doi.org/10.1039/D0CP03867A
Rahman F, Tabrez S, Ali R, Alqahtani AS, Ahmed MZ, Rub A (2021) Molecular docking analysis of rutin reveals possible inhibition of SARS-CoV-2 vital proteins. J Tradit Complement Med 11(2):173–179. https://doi.org/10.1016/j.jtcme.2021.01.006
Deetanya P, Hengphasatporn K, Wilasluck P, Shigeta Y, Rungrotmongkol T, Wangkanont K (2021) Interaction of 8-anilinonaphthalene-1-sulfonate with SARS-CoV-2 main protease and its application as a fluorescent probe for inhibitor identification. Comput Struct Biotechnol J 19:3364–3371. https://doi.org/10.1016/j.csbj.2021.05.053
Al-Karmalawy AA, Soltane R, Abo Elmaaty A, Tantawy MA, Antar SA, Yahya G, Mostafa A (2021) Coronavirus disease (COVID-19) control between drug repurposing and vaccination: a comprehensive overview. Vaccines 9(11):1317. https://doi.org/10.3390/vaccines9111317
Ashour NA, Elmaaty AA, Sarhan AA, Elkaeed EB, Moussa AM, Erfan IA, Al-Karmalawy AA (2022) A systematic review of the global intervention for SARS-CoV-2 combating: from drugs repurposing to molnupiravir approval. Drug Des Dev Ther. https://doi.org/10.2147/DDDT.S354841
Irwin JJ, Tang KG, Young J, Dandarchuluun C, Wong BR, Khurelbaatar M, Sayle RA (2020) ZINC20—a free ultralarge-scale chemical database for ligand discovery. J Chem Inf Model 60(12):6065–6073. https://doi.org/10.1021/acs.jcim.0c00675
O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR (2011) Open Babel: an open chemical toolbox. J Cheminformatics 3(1):1–14. https://doi.org/10.1186/1758-2946-3-33
Eberhardt J, Santos-Martins D, Tillack AF, Forli S (2021) AutoDock Vina 1.2. 0: new docking methods, expanded force field, and python bindings. J Chem Inf Model 61(8):3891–3898. https://doi.org/10.1021/acs.jcim.1c00203
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31(2):455–461. https://doi.org/10.1002/jcc.21334
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30(16):2785–2791. https://doi.org/10.1002/jcc.21256
Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E (2015) GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1:19–25. https://doi.org/10.1016/j.softx.2015.06.001
Grosdidier A, Zoete V, Michielin O (2011) SwissParam: a fast force field generation tool for small 150 organic molecules. J Comput Chem 32:2359–2368. https://doi.org/10.1002/jcc.21816
Vanommeslaeghe K et al (2010) CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem 31(4):671–690. https://doi.org/10.1002/jcc.21367
Zaki AA, Ashour A, Elhady SS, Darwish KM, Al-Karmalawy AA (2022) Calendulaglycoside A showing potential activity against SARS-CoV-2 main protease: molecular docking, molecular dynamics, and SAR studies. J Tradit Complement Med 12(1):16–34. https://doi.org/10.1016/j.jtcme.2021.05.001
Xiong G et al (2021) ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res 49(W1):W5–W14. https://doi.org/10.1093/nar/gkab255
Adasme MF, Linnemann KL, Bolz SN, Kaiser F, Salentin S, Haupt VJ, Schroeder M (2021) PLIP 2021: expanding the scope of the protein–ligand interaction profiler to DNA and RNA. Nucleic Acids Res 49(W1):W530–W534. https://doi.org/10.1093/nar/gkab294
Goyal B, Goyal D (2020) Targeting the dimerization of the main protease of coronaviruses: a potential broad-spectrum therapeutic strategy. ACS Comb Sci 22(6):297–305. https://doi.org/10.1021/acscombsci.0c00058
Narayanan A, Narwal M, Majowicz SA, Varricchio C, Toner SA, Ballatore C, Jose J (2022) Identification of SARS-CoV-2 inhibitors targeting Mpro and PLpro using in-cell-protease assay. Commun Biol 5(1):169. https://doi.org/10.1038/s42003-022-03090-9
Acknowledgements
The author would like to thank Central University of South Bihar Bioinformatics Department and Maulana Azad National Institute of Technology Biological Science and Engineering Department for constant support and technical resources.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
Data collection and output processing were contributed by AM; analysis and interpretation were contributed by AM and SKS; writing was contributed by AM and KMP; critical review was contributed by AM, KMP, and KKO.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
The author declares no conflict of interest.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1. Fig.S1.
Two domains in the SARS CoV-2 main protease Mpro and the interacting residues CYS-145 and HIS-41 forming the catalytic dyad. Fig.S2. Chemical structure of Rutin (Flavonoid). Fig.S4. Grid box placed at the active site residues (HIS-41, SER-46, LEU-141, ASN-142, CYS-145, GLU-166, PRO-168, GLN-189, THR-190, ALA-191) using AutoDock4.2. Table S1. Top 10 Screened FDA Drugs
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mukherjee, A., Pandey, K.M., Ojha, K.K. et al. Identification of possible SARS-CoV-2 main protease inhibitors: in silico molecular docking and dynamic simulation studies. Beni-Suef Univ J Basic Appl Sci 12, 69 (2023). https://doi.org/10.1186/s43088-023-00406-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43088-023-00406-4