
Abstract
Background
Breast cancer is a major form of health problem on the globe and the second cause of death related to cancer amidst women. A prediction of about 1 to 1.3 million cases on cancer of the breast are detected yearly globally. Triple-negative type of breast cancers (TNBCs) are described by the lack of human epidermal growth factor receptor 2 (HER2), estrogen receptor (ER), and progesterone receptor (PR). TNBCs metastasize to the central nervous system and lungs regularly. Such metastatic actions reduce the life expectancy of patients with TNBC than patients with non-TNBC due to non-enhanced inhibitor compounds. The purpose of this research was to explore the anti-proliferative activities of 2-anilinopyrimidine derivatives against triple-negative cancer cell line MDA-MB-468 via in silico studies like QSAR and molecular docking studies to further design and develop new anti-breast cancer drug with high potency and low toxicity.
Results
The quantitative structure–activity relationship QSAR model predicts the bioactivities of the compounds, and molecular docking studies comprehend the interaction between the derivatives (ligand) and thyroid hormone (TRβ1) (receptor). Model 4 was chosen as the best model from the statistical assessment; R2 = 0.8760, R2adj = 0.8451, Q2 = 0.6141, and R2pred of 0.5390. From the external validation of the QSAR model, the coefficient of the mean effect on the model parameters indicates that decreasing (VR1_Dzv and MOMI-R) and increasing (SpMin1_Bh and C3SP3) would increase the anti-proliferative activities (pIC50) of the compounds. The molecular docking studies revealed that ligands 15 and 18 had the highest docking scores of − 7.3 and − 7.4 kcal/mol with thyroid hormone receptor (TRβ1). The ligands had docking scores better than the standard anti-breast cancer drug gefitinib (− 5.3 kcal/mol).
Conclusions
The results indicate that model 4 can be used in developing new 2-anilinopyrimidine derivatives, with better anti-breast cancer prediction activity and performance. It was proved that some series of 2-anilinopyrimidine derivative compounds bind tightly to the receptor, stabilizing the receptor (TRβ1) which is evident from the receptor–ligand interactions, and these compounds would serve as the most promising inhibitors against TRβ1. This shows a breakthrough for pharmaceutical researchers in designing and developing new anti-triple-negative breast cancer drugs.
Similar content being viewed by others

1 Background
After cardiovascular diseases, cancer is the second most deadly disease to the human health [1]. Worldwide, one of seven main death causes is cancer that affects around 14 million people every year. The adoption of lifestyle activities especially in developing countries where almost 82% of the entire population of the world exist has increased higher risk of cancer, due to lack of exercise, smoking, and heredity variation [2]. Breast cancer is the utmost form of cancer on the globe and the second cause of death related to cancer amidst women. A prediction of about 1 to 1.3 million cases on cancer of the breast is detected yearly globally [3].
Triple-negative type of breast cancers (TNBCs) are termed as antagonistic mammary growths, and they are described by the lack of human epidermal growth factor receptor 2 (HER2), estrogen receptor (ER), and progesterone receptor (PR) [4]. TNBCs metastasize to the central nervous system and lung regularly than non-TNBCs, which usually metastasize to the bone. Such metastatic actions reduce the life expectancy of patients with TNBC than patients with non-TNBC patients due to non-enhanced inhibitors compounds [3].
Recently, a novel series of 2-anilinopyrimidine was reported by Jo et al. [4] as inhibitors against MDA-MB-468 cell line. There is also evidence that reduced thyroid hormone receptor manifestation and/or variations in thyroid hormone genes occur frequently in cancer [5], suggesting that the native receptors could act as tumor suppressors and that loss of re-occurrence of this receptor could show a selective lead for cell alteration and advancement of tumor transformation [6].
Unconventional medicine takes prolonged time and effort to be manufactured, thereby not meeting up with the urgency needed for a comprehensive treatment. Computer-aided drug design has been a great success in designing novel drugs with great effectiveness and better potency against diseases. The aim of this study is to explore the anti-proliferative activities of 2-anilinopyrimidine against triple-negative cancer cell line, MDA-MB-468 via in silico studies like QSAR and docking studies that can be used to further develop anti-breast cancer drug candidate.
2 Methods
2.1 Computational information
2.1.1 Hardware
The computer details used in this research is the 7th Generation HP Pavilion Intel R, core i7-7500u RAM 12.00 GB running on a Windows 10 operating system.
2.1.2 Software
The software used to carry out this research includes Spartan’14 (version 1.1.2), Material studio (V8), AutoDock visualiser version 4.2, Pyrex software version, ChemDraw software version 12.0.2, PADEL-Descriptor Software V2.20 and DTC data lab software, and Microsoft Word Office Excel 2013 version.
2.2 QSAR studies
2.2.1 Data collection
Thirty novel derivative compounds of 2-anilinopyrimidine with their inhibitory concentration (IC50), against triple-negative breast cancer (MDA-MB-468) cell line, were acquired from Jo et al. [4] reports.
2.2.2 Bioactivities
Anti-proliferative activities of 2-anilinopyrimidine derivative compounds were measured in inhibitory concentration (IC50); an IC50 (50% inhibitory concentration) value of a chemical compound is defined as the concentration of the compound required to decrease the viability of a given cell line by 50%. The IC50 values were normalized using the scale of logarithm to pIC50 values to reduce the skew in the activities. The tabulated anti- proliferative activities (IC50) and pIC50 of the derivatives are shown in Table 1, and it is measured in the concentration of micromolar (μM). The logarithm scale is given as follows:
pIC50 = − log10 (IC50 × 10−6).
2.2.3 Geometry optimization
The geometry optimization is aimed to earn a more desirable geometric structure that is closer to the actual geometric condition of the molecular structure [2]. The derivative compounds were sketched in 2D on ChemDraw V (12.0.2) and converted on Spartan 14 V (1.1.4) software. Density functional theory (DFT) using the B3LYP, 6-311G basis set, was used for the geometric optimization of the compounds [7,8,9]. The parent compound is shown in Fig. 1.

Parent compound of 2-anilinopyrimidine derivatives
2.2.4 Molecular descriptor
Pharmaceutical Data Exploration Laboratory Software V (2.20) was used in calculating molecular descriptors for the 30 optimized compounds of 2-anilinopyrimidine derivatives [10].
2.2.5 Pretreatment and division of data set
Results obtained from PADEL-software were pretreated using Data Pre-treatment software GUI 1.2 to remove constant values and unwanted descriptors [9, 11]. Kennard-Stone algorithm [12] was used in dividing the derivatives into 21 train and 9 test set to build the model.
2.2.6 Model building and model validation
The internal validation of the train test (twenty-one compounds) was executed in version 8 of Material studio software to construct a model by employing a genetic function approximation technique. Using the Friedman formula, the obtained models were evaluated [13].
where SEE is the standard estimated error. If SEE is low, it implies a better model. SEE is expressed as follows:
C is the sum of the model terms, p is the total number of model descriptors, M is the sum of train set, and d is a user-defined smoothing parameter [14]. The model is verified using the correlation coefficient (R2). R2 value should be close to 1, to obtain an enhanced and effective model. R2 is given as follows:
where Yexp and Ypred are averages of anti-proliferative and predicted activities of the train set [15].
R2 value increases as the descriptor number increases; thus, R2 value is not guaranteed in terms of the model’s strength. The R2 is altered to obtain a robust and strong model, which is given as follows:
where p and n are the numbers of generated descriptors and train set. The stability of the model derivatives was assessed using validation coefficient test (Q2cv) given as:
Ytraining, Yexp, and Ypred are the average biological activities (pIC50), biological activities (pIC50), and prediction inhibition concentration of the train set [16].
2.2.7 QSAR modeling evaluation
The models generated were set to undergo statistical parameters such as the cross-validated test, R2 Fisher’s test, and R2 predicted.
2.2.8 Mean effect
The mean effect relates to the impact of the descriptors and the compound activities in the model. Notations attached to the descriptors show the variant direction in the activity of the compounds, either an increase or a decrease in the descriptor value. It is defined as follows:
where m is the total descriptors in the model, Bj equals to descriptor coefficient j, n is the total molecules in the train set, and Dj is the matrix value of the descriptor in the train set [17].
2.2.9 Variance inflation factor (VIF)
The VIF measures the extent of correlation between one descriptor and the other descriptor in a model. The higher the values show that it is almost impossible and difficult to show the contribution of a descriptor accurately in a model. It is evaluated as follows:
R2 is the correlation coefficient [18].
The higher the value, the greater the correlation between the descriptors. Values of 1–7 are sometimes regarded as being moderate, and it shows the strength and robustness of the model, while values of 10 show the correlation between the descriptors is very high, and therefore, the model is very unstable.
2.2.10 QSAR applicability domain of the model
The goal of applicability domain methods is for estimating individually the consistency of each model generated [19], and it aimed at predicting the uncertainty of a compound depending on its similarities to the compounds used in building the model and also the distance of both train and test set. The leverage is used in defining the applicability domain of the generated models [20]. It is formulated as follows:
where X is the n × k matrix of train set descriptors, XT is the matrix transpose of X used in building the model, and Xi is the matrix of train compounds of I. (h*) is the warning leverage, and it is a prediction tool that checks for outliers. It is written as follows:
n is equal to the total train set and p equals to the total descriptors from the model generated. William’s plot is generated by plotting the standardized residuals versus the leverage of both the train and test set. Molecules that fall within the warning leverages on the plot are the predicted compounds that fall within the threshold. The reliability of the QSAR model was assessed using the minimum accepted values as shown in Table 3 [21].
2.3 Molecular docking
Molecular docking studies were implemented on the derivative compounds of 2-anilinopyrimidine (ligand) and thyroid hormone receptor (TRβ1). The receptor was gotten from protein data bank with the code (PDB: 1Y0X). The docking scores of the ligand–receptor were obtained with Autodock Vina of PyRx software [11]. The detailed interactions between the ligand and the receptor were visualized using Discovery Studio Visualizer.
3 Results
3.1 QSAR of 2-anilinopyrimidine derivatives
Four QSAR models were generated using the Genetic Function Approximation (GFA) technique to predict the anti-proliferative activities. Model 4 passed the internal validation test which confirmed with the least requirement for QSAR modeling as shown in Table 2.
3.1.1 Model 1
pIC50 = − 0.000041993 × VR1_Dzv + 0.430022665 × C3SP3 − 0.029366849 × RDF125m − 0.013797643 × RDF105p + 4.414124338
3.1.2 Model 2
pIC50 = 0.019329185 × ALogp2 + 0.209407843 × C3SP3 + 0.013676289 × RDF40i − 0.000911095 × Vm + 3.736702488
3.1.3 Model 3
pIC50 = − 0.015741625 × VR3_Dzv + 0.385603503 × C3SP3 − 0.036977855 × RDF125m − 0.016463267 × RDF105p + 4.720613746
3.1.4 Model 4
pIC50 = − 0.000060824 × VR1_Dzv + 1.185723768 × SpMin1_Bhs + 0.378178925 × C3SP3 − 0.128667903 × MOMI-R + 3.282331294
Tables 3 and 4 show the calculation of the external validation of the QSAR model using the model parameters of model 4. The external validation (R2pred) was calculated as 0.5390, which also conforms to the minimum required values for QSAR modeling, and makes the model very robust and highly potent. The meaning of each model parameter used in validating model 4 is given in Table 6.
The experimental, predicted, and the residual values of 2-anilinopyrimidine derivatives are shown in Table 5. The residual values were obtained from the calculated activities statistically. All the derivative compounds had low residual values indicating the degree of effectiveness of the QSAR model 4.
Table 6 shows the four model parameters (descriptors) that were used in building the QSAR model 4 and were also used in evaluating the strength of the model externally. The descriptors are defined and classified in Table 6.
Table 7 shows the statistical evaluation (VIF, mean effect, P values) of the model parameters. The VIF shows the degree of co-linearity between the descriptors, and it was calculated using the following equation:
R2 is the correlation coefficient [18].
The mean effect shows the contribution of each descriptor to the built model, and the signs of the values show if the descriptors give a negative or positive contribution in the model. The P values evaluate the statistical significance between the model parameters.
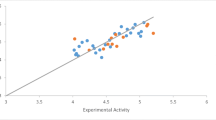
Figure 2 shows a straight line graph of calculated activities (predicted activities) against experimental activities of 2-anilinopyrimidine derivative compounds as tabulated in Table 5. Both the experimental and predicted activities showed a good relationship as proven by the graph.

Graph of predicted activities versus experimental activities (bioactivities)
Figure 3 shows a graph of standardized residual against inhibition concentration of both the train and test set. All the values were well distributed on both sides of the y-axis, showing the effectiveness of model 4.

Graph of standardized residual against inhibition concentration (experimental activity)
Figure 4 is a graph of standardized residuals against the leverage values, and the plot is called William’s plot. The plot was used to assess the uncertainty in similarities of the derivative compounds used in building the model. Compounds that fall between the warning leverage tend to be similar structurally. The warning leverage was calculated to be (h* = 0.714) using the formula:

A plot of standardized residual versus leverages (William’s plot)
3.2 Molecular docking studies
The summary of the docking studies result of some 2-anilinopyrimidine derivative compounds is given in Table 8. The docking score was obtained using PyRx software while the docking interactions between the receptor and the ligand to form complexes which include hydrophobic bond, hydrogen bond, and the bonding distances were visualized using Discovery Studio Software. The hydrogen and hydrophobic interaction that occurred between 2-anilinopyrimidine derivative compounds (ligand) and the active pocket of (TRβ1) receptor in 3D format for complexes 15 and 18 are shown in 2D format in Figs. 6 and 7, while Fig. 8 shows the same interaction in a 3D format.
4 Discussion
4.1 QSAR of 2-anilinopyrimidine
QSAR modeling was used to validate quantitatively the structure relationship of 2-anilinopyrimidine derivatives with its anti-proliferative activities. The robustness of the QSAR models was assessed by the fitness of the train set and predicted pIC50 of the test set. Four QSAR models generated using the Genetic Function Approximation (GFA) technique to predict the anti-proliferative activities. Model 4 passed the internal validation with correlation coefficient squared (R2) of 0.8760, correlation coefficient adjusted squared (R2adj) of 0.8451, cross-validation coefficient (Q2) of 0.6141, and external validation (R2pred) of 0.5390. All the values obtained were in accordance with the least proposed value used in the evaluation of QSAR model as shown in Table 2. The obtained values (R2, R2adj, Q2, and R2pred) indicate the existence of a high correlation between the predicted pIC50 along with the biological pIC50 of the data set.
4.1.1 External validation of QSAR model 4
Model 4 was verified as the best model using the descriptors from the test set of the derivative compounds. Tables 3 and 4 show how the external validation was achieved using the values of the descriptors from the test set. The experimental, predicted, and the residual values of 2-anilinopyrimidine derivatives are shown in Table 5. The low residual value from biological (anti-proliferative) activities and predicted activities shows the high performance of the model.
Table 6 shows the definition of the descriptors (model parameters). The mean effect result (Table 7) showed the degree of impact of each descriptor on the model, and the values and coefficients of the descriptors show that decreasing MOMI-R and then VR1_Dzv (negative descriptors) would increase the anti-proliferative activities of the derivative compounds while increasing SpMin1_Bh followed by C3SP3 (positive descriptors) which would also increase the anti-proliferative activities of 2-anilinopyrimidine derivative compounds. The variance inflation factor (VIF) showed that there is no much inter-correlation between the descriptors making the model very stable. The null hypothesis shows no significant connection amid the bio-activity and model parameters of the constructed model at p > 0.05. At 95% confidence level, the P values of the model parameters were below 0.05. Therefore, the null hypothesis is rejected and the alternative hypothesis is accepted as shown in Table 7.
Figure 2 shows the plot of predicted activity (pIC50) versus the experimental activity (IC50) of both the test set and train set of compounds. The plot showed that the predicted activity was in good agreement with its experimental values as shown in Table 2, conforming the effectiveness and strength of the built model. Figure 3 is a plot of standardized residual versus biological activity (inhibition concentration) of both the train set and test set, and it shows the values spread on both sides of the zero point of the plot, showing no systematic errors. Figure 4 is a graph of standardized residuals against the leverage value, and the plot is called William’s plot. Almost all the compounds were within the calculated warning leverage (applicability domain) of h* = 0.714, and compounds 2, 8, 15, and 14 were found to be outside the warning leverage which perhaps is because of the slight difference in their structures equated to other molecules of the data set. Both internal and external validation conform model 4 to be very stable, robust, and highly predictive.
4.2 Molecular docking studies
Molecular docking studies on compounds of 2-anilinopyrimidine with the protein target thyroid hormone receptor (TRβ1) were performed. Amongst all the derivatives, compounds 12, 15, 18, and 30 had high docking scores. The prepared receptor and ligand are shown in Fig. 5. Compounds 15 and 18 had the highest docking score of – 7.4 and – 7.3 kcal/mol as shown in Table 8. The visual examination of the docked complexes was done by evaluating the hydrogen bond interaction, hydrogen bond length, and hydrophobic interaction.

3D structure of the ligand and prepared protein receptor
Compound 15 showed the backbone conventional hydrogen bonding interaction with ARG 429 (2.50 A0) and two amino acids of GLU311 (2.7609 A0 and 2.1551 A0). Again, VAL458 showed carbon–hydrogen interaction with the compound at distance of 3.3765 A0. Also, the pi-orbital containing delocalized electrons in the benzene ring interact with the alkyl groups of ILE303 (5.4379 A°), LYS306 (5.04683 A°), and ARG383 (5.3858 A°) and three amino acids of PRO384 (5.1107 A°, 4.7845 A°, and 4.7531 A°) to form hydrophobic bond.
Compound 18 also showed the same hydrogen bonding with amino acid residues of GLU311 (2.10982 A°), ARG439 (2.68544 A°), GLY307 (2.97669 A°), GLU311 (2.85424 A°), and carbon–hydrogen bonding with VAL458 (3.34145 A°). Furthermore, the benzene ring of the compound interacts with the alkyl groups the amino acid residues formed hydrophobic bond, and they include ILE303 (5.28774 A°), LYS306 (4.9622 A°), ARG383 (5.40494 A°), PRO384 (4.84454 A° and 5.15235 A°), and ALA436 (4.91503 A°).
Both compounds were adequately docked and their orientation is similar in some instances, validating the good quality of the docking results. Both compounds showed the same hydrogen bond and hydrophobic bond interactions with the amino acid residues of the receptor at different distances. The ligands had docking scores better than the standard drug gefitinib (− 5.3 kcal/mol). From the compound interaction with the receptor, it proves the ability of the compounds to inhibit TRβ1 receptor. Figures 6 and 7 give detailed binding interactions of the receptor with ligands 15 and 18 while Fig. 8 shows how the ligand (compound) binds firmly to the active site of the protein receptor to form complexes in 3D with ligands 15 and 18.

Receptor–ligand interaction of complex 15 on a 2D diagram

Receptor–ligand interaction of complex 18 on a 2D diagram

Ligand interactions of complexes 15 and 18 in a 3D diagram
5 Conclusion
2-Anilinopyrimidine derivatives were proven to be a better anti-cancer drug candidate against MDA-MB-468 cell line from both QSAR studies and molecular docking studies that were carried out to predict a better activity from the experimental activity of the derivatives and also comprehend the interaction of the ligand (derivatives) and thyroid hormone receptor (TRβ1). The coefficient and values of the mean effect of QSAR model 4 indicate that increasing Spmin1_Bhs and C3SP3 descriptors will increase the anti-proliferative activities of the derivatives while decreasing VR1-DZv and MOMI-R descriptors would also increase the activities of 2-anilinopyrimidine derivatives as a standard anti-breast cancer agent. The robustness, applicability, and predicted capacity of the model generated were analyzed for both internal and external validation test which conforms to the minimum recommended values. This indicates that model 4 can be used in developing new 2-anilinopyrimidine derivative compounds with better anti-breast cancer activity. The molecular docking result showed that compounds 15 and18 had the highest docking score of − 7.4 and − 7.3 kcal/mol, when it is compared to the standard drug gefitinib. From the studies, it is proven that some series of 2-anilinopyrimidine derivative compounds bind tightly to the receptor, stabilizing the receptor (TRβ1) which is evident from the receptor–ligand interactions. The compounds would serve as the most promising inhibitors against TRβ1. This research would be a breakthrough for pharmaceutical researchers in designing and developing new anti-triple-negative breast cancer drugs.
Availability of data and materials
Not applicable
Abbreviations
- QSAR:
-
Quantitative structure-activity relationship
- TNBCs:
-
Triple-negative breast cancers
- TRβ1:
-
Thyroid hormone receptor
References
Lan J, Huang L, Lou H, Chen C, Liu T, Hu S et al (2018) Design and synthesis of novel C14-urea-tetrandrine derivatives with potent anti-cancer activity. European journal of medicinal chemistry 143:1968–1980
Putri, D. E., Pranowo, H. D., & Haryadi, W. I. N. A. R. T. O. (2019). Study on anti-tumor activity of novel 3-substituted 4 anilino-coumarin derivatives using quantitative structure-activity relationship (QSAR). In Materials Science Forum (Vol. 948, pp. 101-108). Trans Tech Publications.
Ge W, Hao X, Han F, Liu Z, Wang T, Wang M et al (2019) Synthesis and structure-activity relationship studies of parthenolide derivatives as potential anti-triple negative breast cancer agents. European journal of medicinal chemistry 166:445–469
Jo J, Kim SH, Kim H, Jeong M, Kwak JH, Han YT et al (2019) Discovery and SAR studies of novel 2-anilinopyrimidine-based selective inhibitors against triple-negative breast cancer cell line MDA-MB-468. Bioorganic & medicinal chemistry letters 29(1):62–65
Aranda A, Martínez-Iglesias O, Ruiz-Llorente L, García-Carpizo V, Zambrano A (2009) Thyroid receptor: roles in cancer. Trends in Endocrinology & Metabolism 20(7):318–324
Martínez-Iglesias, O., Ruiz-Llorente, L., Jurado, C. C., & Aranda, A. (2014). Thyroid hormone receptors and their role in cell proliferation and cancer. In Cellular Endocrinology in Health and Disease (pp. 1-17). Academic Press.
Abdullahi M, Uzairu A, Shallangwa GA, Mamza P, Arthur DE, Ibrahim MT (2019) In-silico modelling studies on some C14-urea-tetrandrine derivatives as potent anti-cancer agents against prostate (PC3) cell line. Journal of King Saud University-Science
Adeniji SE, Uba S, Uzairu A (2018) Quantitative structure–activity relationship and molecular docking of 4-alkoxy-cinnamic analogues as anti-mycobacterium tuberculosis. J King Saud University-Science
Arthur DE, Uzairu A, Mamza P, Abechi SE, Shallangwa G (2018) Activity and toxicity modelling of some NCI selected compounds against leukemia P388ADR cell line using genetic algorithm-multiple linear regressions. J.King Saud University-Science
Yap CW (2011) PaDEL-descriptor: an open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 32:1466–1474
Abdulfatai U, Uzairu A, Uba S (2018) Molecular docking and quantitative structure-activity relationship study of anticonvulsant activity of aminobenzothiazole derivatives. Beni-Suef University Journal of Basic and Applied Sciences 7(2):204–214
Kennard RW, Stone LA (1969) Computer aided design of experiments. Technometrics 11:137–148
Friedman JH (1991) Multivariate adaptive regression splines. Ann. Stat. 1–67
Khaled KF, Abdel-shafi NS (2011) Quantitative structure and activity relationship modeling study of corrosion inhibitors: genetic function approximation and molecular dynamics simulation methods. Int. J. Electrochem. Sci. 6:4077–4094
Tropsha A, Gramatica P, Gombar VK (2003) The importance of being earnest: validation is the absolute essential for successful application and interpretation of QSPR models. Mol. Inform. 22:69–77
Brandon V., Orr, A., 2015. Comprehensive R archive network (CRAN): http://CRAN.Rproject.org.
Minovski N, Zˇuperl Š, Drgan V, Novicˇ M (2013) Assessment of applicabilitydomain for multivariate counter-propagation artificial neural network predictive models by minimum Euclidean distance space analysis: a case study. Anal. Chim. Acta 759:28–42
Myers RH (1990) Classical and modern regression application. Duxbury Press, CA
Eriksson L, Jaworska J, Worth AP, Cronin MT, McDowell RM, Gramatica P (2003) Methods for reliability and uncertainty assessment and for applicability evaluations of classification-and regression-based QSARs. Environmental health perspectives 111(10):1361–1375
Veerasamy R, Rajak H, Jain A, Sivadasan S, Varghese CP, Agrawal RK (2011) Validation of QSAR models-strategies and importance. Int.J.DrugDes.Discov 3:511–519
Ibrahim MT, Uzairu A, Shallangwa GA, Ibrahim A (2018) In-silico studies of some oxadiazoles derivatives as anti-diabetic compounds. J King Saud University-Science
Acknowledgements
The authors acknowledge the technical effort of the Chemistry Department, Ahmadu Bello University, Zaria, for their enormous contribution and guidance toward the successful completion of this research article.
Consent of publication
Not applicable
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
HAL gathered the data set and analyzed it computationally in accordance with the methodology to get a model of high performance and was a major contributor in drafting the manuscript. AU carried out the statistical analysis on the model to ensure its stability and also participated in the write up, and SU re-edited the work and ensured it conforms with the manuscript’s guide. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abdulrahman, H.L., Uzairu, A. & Uba, S. In silico studies of some 2-anilinopyrimidine derivatives as anti-triple-negative breast cancer agents. Beni-Suef Univ J Basic Appl Sci 9, 13 (2020). https://doi.org/10.1186/s43088-020-00041-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43088-020-00041-3