
Abstract
Background
Bardet-Biedl syndrome (BBS) is a rare multisystemic autosomal recessive (AR) disorder, which falls under the spectrum of ciliopathic disorders. As BBS is a very rare entity in India, its diagnosis is most often missed during early child visits. The lack of a syndromic approach for diagnosing genetic disorders by health care physicians is being considered a major blackguard. The following case report exemplifies how a patient presenting with multisystemic involvement should be evaluated to rule out syndromic association.
Case presentation
The authors here report a case of a male child aged 13 years presenting to Pediatrics Outpatient with complaints of learning disability and behavioral disturbances. During his initial assessment, features such as polydactyly, overweight, and vision disturbances were picked up by the pediatrician as an indication towards syndromic association. Soon a complete laboratory workup and various scans were done which revealed hepatic fibrosis and gonadal dysgenesis. Simultaneously, IQ testing was recommended which was suggestive of mild mental retardation. Bringing along all these clinical presentations a diagnosis of BBS was made. Post-diagnosis parents were counseled on recurrence risk and explained the importance of regular follow-ups and screening to improve quality of life.
Conclusion
This case report emphasizes the role of holistic multidisciplinary approach for diagnosing at early stage and better prognosis of BBS. Prenatal genetic counseling along with next-generation sequencing are a few potential measures to drop the incidence of this condition. Obesity and visual disturbances are a few concerns which if not handled early can result in unfortunate outcomes. Renal involvement in BBS is considered a deadly parameter which surely was not seen in this case. For all learning/intellectual disabilities, the triad of screening, clinical examination, and interdisciplinary approach can clinch in early diagnosis of a genetic syndrome.
Similar content being viewed by others


Background
The Bardet-Biedl syndrome (BBS) is a rare multisystemic autosomal recessive (AR) disorder, which falls under the spectrum of ciliopathic disorders. The frequency of the syndrome is estimated to be 1:160,000 [1]. However, the incidence is much higher in populations with a higher level of consanguinity and some isolated human communities. Diagnosis of BBS is well established by clinical manifestations including primary features (Rod-Cone dystrophy, polydactyly, obesity, learning disabilities, genital abnormalities, renal anomalies) and secondary characteristics (speech disorder, developmental delay, brachydactyly, strabismus/cataract/astigmatism, diabetes mellitus, ataxia, polyuria/polydipsia, dental defects, hepatic fibrosis, left ventricular hypertrophy); presence of [3 primary + 2 secondary] OR [4 primary] features confirm the diagnosis [1]. At least 21 BBS genes with unique mutations have already been identified which play a pivotal role in ciliary function [2]. BBS is more of a clinical diagnosis rather than genetic detection. This case report emphasizes the role of detailed clinical examination along with next-generation sequencing for early diagnosis and better prognosis of BBS.
Case presentation
Our Proband, a 13-year-old boy, was the first son of a young (mother 33 years old, father 34 years old) and an apparently healthy couple. The family self-reported as non-consanguineous. The second son (9 years old) was completely normal. The patient presented to Pediatrics Outpatient 6 months back with complaints of decreased intelligence as compared to children of the same age group, inability to understand and follow one/two-step commands, aggressive and self-harming behavior, hyperactive and inattentive in school, abnormal eating behavior (asking repeatedly for food), and addicted to phone.
Anthropometry revealed that his BMI was 21.7 kg/m2 (> 1 standard deviation [SD] above the median according to WHO, BMI-for-age-BOYS z-scores), with a weight of 33.6 kg and height of 124.5 cm (< − 3 standard deviation [SD] according to WHO, height-for-age z-scores). On examination, the patient was conscious, non-cooperative, and oriented to the mother. He was hemodynamically stable and his vitals were as follows: heart rate—90/min; respiratory rate—20/min; BP—50 to 90th percentile; SpO2—98%.
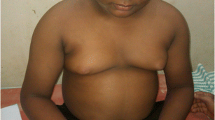
When head-to-toe examination was performed, we found that patient has microcephaly with a head circumference of 43 cm (< − 3 standard deviation [SD] according to WHO, head circumference-for-age z-scores). Several dysmorphic traits (as described in Fig. 1) were also observed namely a narrow forehead, sparse eyebrow hypertelorism, almond-shaped eyes, broad nasal bridge, smooth philtrum, post-axial polydactyly (as described in Fig. 2). An ophthalmological fundus examination revealed signs of macular degeneration (as described in Fig. 3); on further questioning, parents mentioned that patient’s night vision was also very poor (banging his head on the wall while walking).

Image of the child at 12 years of age showing several dysmorphic traits namely a narrow forehead, sparse eyebrow hypertelorism, almond shaped eyes, broad nasal bridge and smooth philtrum

Figure-2 depicts postaxial hexadactyly (an accessory digit on the ulnar side of the left hand)

Fundus photo of the right eye representing features suggestive of macular degeneration
The neurological evaluation revealed normal tone and a power of 5/5 in all four limbs. Higher Mental Functions testing suggested childhood speech language disorder due to cognitive impairment. Superficial and deep tendon reflexes were normal. MRI brain was performed and no significant neuro parenchymal abnormality was detected.
Psychological assessment revealed behavioral disturbances like self-harming nature, excessive aggressiveness (pulling his mothers’ hair or hitting younger sibling), hyperactive (cannot sit at a place for more than 15 min), and inattentive (cannot follow commands), needing help with all day-to-day activities including using washroom, bathing, or dressing/undressing. The child was diagnosed with mild mental retardation due to an I.Q. of 52 (according to WHO’s ICD-10 diagnostic criteria for mental retardation). Patient was also asked to get a 2D ECHO and color Doppler impression of which was normal.
Per abdomen examination revealed central obesity and on deep palpation hepatomegaly was suspected which was confirmed by USG abdomen with a liver span of 16.1 cm (about 6.4 in.), firm in consistency, and rounded border. Liver function tests were deranged, as mentioned in Table 1. So, a Fibro scan/elastography was suggested by gastro medicine department which gave a diagnosis of moderate hepatic fibrosis F2 (Stiffness- 9.9kpa). Post-scan, a liver biopsy was planned and done. Results of which are as follows: mild portal widening with moderate mononuclear inflammation. Hepatocytes show ballooning degeneration, micro and macro vesicular fatty changes and mild intrahepatic cholestasis. The final impression was of a non-alcoholic fatty liver disease [Grade 1]. Measurements of spleen, right and left kidney were all found normal on USG.
The patient was also assessed by the endocrinological department where he was suspected of having subclinical hypothyroidism (no history of any thyroid disorder in the family) based on altered thyroid function tests, as mentioned in Table 1. Along with that several hormonal levels (LH, Testosterone) were also checked (levels mentioned in Table 1) as the genitalia examination pointed towards micro-orchidism with stretched penile length of 4 cm (7.1 ± 1.6 cm). As IGF-1 levels were found to be low (54.2 ng/ml), it was followed by a GH stimulation test. Results of which were found to be normal interpreting this as primary IGF deficiency due to GH resistance.
Audiological testing was done due to complaints of bilateral decreased hearing and intermittent pus-like discharge since childhood (CSOM, Safe/inactive type) via brain evoked response auditory (BERA) test which confirmed the diagnosis of bilateral moderate conductive hearing loss due to delayed absolute latencies.
After a detailed developmental history, a major lag [around 3 years] in achieving developmental milestones (language > social > fine and gross motor) was recognized and it was of the impression that the boy has global developmental delay with development quotient of 46.15 (severe delay).
As this syndrome has no cure, the plan of management was based on frequent follow-up on a multidisciplinary approach to keep a check on worsening of symptoms and, if so, plan for their early management. Starting with growth velocity monitoring, yearly checks to look for appropriate increase in height are done and if the rates are found to be decreasing further then interventions will be planned accordingly. As the IQ of the boy was found to be low, it was recommended to place him in school for Special Education. Annual fibro scans along with LFTs were planned to look for worsening of liver pathology and keep a check on liver functions. Currently, the child has no complaints of difficulty in near/far vision; thus, annual screening tests to detect visual acuity and progression of retinal degeneration (if any) were decided. Obesity-induced health outcomes and renal function deterioration are the two deadly parameters and thus 6-monthly follow-up for the same was considered. Parental counseling played a crucial part in this management plan.
Discussion
Bardet-Biedl syndrome [Mendelian Inheritance in Man,209900] previously considered a part of Laurence-Moon-Biedl-Bardet syndrome is addressed as a separate entity now [3]. The most common gene mutated in this syndrome is BBS1, followed by BBS10. To date, no therapy pertaining to the prevention of progressive deterioration of features of this condition exists [4]. Manifestations of renal disease are variable in BBS, but renal involvement is the leading cause of early death in this condition. In our patient, there was no renal involvement owing to better prognosis.
Retinal dysfunction in BBS is well recognized with the most common trait being retinal dystrophy along with optic atrophy deteriorating vision [5]. Evidence of macular degeneration findings in our case was in correspondence with the fact that 93% of BBS patients exhibit retinal degeneration. In accordance with previous studies, there are no approved treatments to alleviate vision deterioration and prevent blindness; however, regular ophthalmological follow-up is utmost necessary for visual prognosis. Use of low-vision aids can optimize their sight by preserving the remaining vision leading to greater independence in life.
Puberty specifically in males is considered to be a stressful period for those with BBS. This child has delayed puberty due to signs such as short stature, micro-orchidism, shortened stretched penile length, and lagging of features according to age-appropriate Tanner staging of sexual development. Hormonal assays revealed comparatively low total testosterone levels, 0.2 nmol/L. Management involves complete clinical examination, anthropometry, and growth velocity monitoring along with regular check-ups and assessment of hormone (LH, testosterone) levels.
Postaxial polydactyly is the only surest sign of BBS that can be present at birth while other features keep evolving throughout the entire life. Incidence of both upper and lower extremities being simultaneously involved is seen in 21% of patients, lower limb involvement occurring in 21% of the patients and upper limb involvement occurring in 9% of the patients [6]. In our patient, we observed postaxial hexadactyly (an accessory digit on the ulnar side of the left hand). X-ray hand was done to confirm whether it is polydactyly or simply a tissue overgrowth (ex-lipoma/cyst). Accessory digits are often non-functional and may be excised.
Overweight/obesity is a highly prevalent phenotype of BBS and according to previous studies it has come to notice that intrauterine growth and birth weight are duly normal in these patients, but they document early onset of significantly raised BMI z-scores which persists throughout the adolescent period [7]. Most of the cases of BBS report for the first time with complaints of excessive weight gain and hyperphagic behavior. In our case, the patient had overweight, severe stunting, and a deranged lipid profile. This spectrum is a detrimental risk factor for major cardiovascular, liver diseases and autoimmune conditions [8]. Therefore, it is of prime importance that healthcare providers should counsel parents regarding early lifestyle modifications. A low-calorie and low-protein diet helps in obesity control and may also slow the progression of renal failure in patients with BBS [9].
Global developmental delay is a major concerning feature in our case report as this may lead to hampered quality of life. Developmental surveillance, language stimulation activities, and behavioral interventions (ex-psychologist) are a few of the measures that can improve the outcome if implemented early [10]. IQ testing and cognitive assessment help in the proper placement of child to privileged students’ education school. Counseling of family to help the child in leading a normal life still stays at forefront of care planning even after the advent of all these measures.
In this case report, a homozygous BBS10 (ENST00000650064.2), c.271dup (p. Cys91LeufsTer5) variant, was identified using Clinical Whole Exome Sequencing (WES). This mutation was categorized as likely pathogenic based on American College of Medical Genetics and Genomics (ACMG) guidelines and criteria [PMID: 25741868]; [11]. BBS10 is located on chromosome 12q21 and contains two exons with a total length of 23.97 Kb, which codes for a protein of 723 amino acids that belongs to a Chaperonin-like complex [12]. A total of 115 variants distributed across the whole BBS10 gene have been reported in the Human Gene Mutation Database (HGMD) [13]. After a detailed analysis, it has come to notice that approximately 16% of BBS cases results from mutation in BBS10 [14].
There is clearly a major delay in diagnosing BBS in younger patients. This is probably because of both a lack of medical awareness of the condition and the difficulty of making a firm diagnosis owing to the slow emergence of features amongst the constellation of symptoms. The patient was repeatedly followed up and was given varied treatments based on individual clinical features but was never approached on a multidisciplinary basis until today. This delay highlights the importance of complete clinical examination for syndromic diagnosis. Genetic diagnosis of BBS is complicated due to the lack of gene-specific disease symptoms; however, it is gradually becoming more accessible with the invention of multigene sequencing technologies. Such a targeted sequencing provides molecular confirmation in 80% of cases.
Conclusion
A coordinated interdisciplinary approach is required for effective evaluation and betterment of treatment in such pleiotropic condition. Genetic counseling, prenatal testing, evaluation in a holistic manner, and complete hematological workup play a pivotal role in the diagnosis of genetic disorders. Characteristics such as faltering height, developmental delay, and birth abnormality are alarming signs which invoke pediatricians to undergo detailed clinical examination keeping in mind a possible differential of syndromic diagnosis. Regular screening for midline abnormalities should also be recommended whenever a physician is dealing with genetic syndromes. As there is no known remedy for BBS, the major need for early implementation of treatment is improving quality of life, decreasing mortality and morbidity rates, obesity control, and better visual prognosis. Parents should be counseled on recurrence risk and called for regular follow-up to observe whether there are any progressive changes and for behavioral therapy. For all learning/intellectual disabilities, the triad of screening, clinical examination, and interdisciplinary approach can clinch in early diagnosis of genetic syndrome.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- BBS:
-
Bardet-Biedl syndrome
- AR:
-
Autosomal recessive
- USG:
-
Ultrasonogram
- BMI:
-
Body mass index
- BP:
-
Blood pressure
- SD:
-
Standard deviation
- IQ:
-
Intelligence quotient
- SGOT:
-
Serum Glutamic Oxaloacetic Transaminase
- SGPT:
-
Serum Glutamic Pyruvic Transaminase
- WES:
-
Whole-exome sequencing
- ACMG:
-
American College of Medical Genetics and Genomics
- HGMD:
-
Human Gene Mutation Database
- RFT:
-
Renal function test
- LFT:
-
Liver function test
- TFT:
-
Thyroid function test
- MRI:
-
Magnetic resonance imaging
- BERA:
-
Brain evoked response auditory
- CSOM:
-
Chronic suppurative otitis media
- WHO:
-
World Health Organization
- ICD:
-
International Classification of Diseases
References
Forsythe E, Beales PL (2013) Bardet-Biedl syndrome. Eur J Hum Genet 21(1):8–13. https://doi.org/10.1038/ejhg.2012.115. Epub 2012 Jun 20. PMID: 22713813; PMCID: PMC3522196
Ohto T, Enokizono T, Tanaka R, Tanaka M, Suzuki H, Sakai A, Imagawa K, Fukushima H, Fukushima T, Sumazaki R, Uehara T, Takenouchi T, Kosaki K (2017) A novel BBS10 mutation identified in a patient with Bardet-Biedl syndrome with a violent emotional outbreak. Hum Genome Var 10(4):17033. https://doi.org/10.1038/hgv.2017.33. PMID:28808579;PMCID:PMC5550758
Hrynchak PK (2000) Bardet-Biedl syndrome. Optom Vis Sci 77(5):236–243. https://doi.org/10.1097/00006324-200005000-00010. PMID: 10831213
Forsyth RL, Gunay-Aygun M. Bardet-Biedl Syndrome Overview. 2003 Jul 14 [Updated 2023 Mar 23]. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews®. Seattle (WA): University of Washington, Seattle; 1993–2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1363/
de Oliveira Andrade LJ, Andrade R, França CS, Bittencourt AV (2009) Pigmentary retinopathy due to Bardet-Biedl syndrome: case report and literature review. Arq Bras De Oftalmol 72(5):694–696. https://doi.org/10.1590/S0004-27492009000500019
Li H, He J, Leong I, Huang R, Shi X (2022) Central precocious puberty occurring in Bardet-Biedl syndrome-10 as a method for self-protection of human reproductive function: a case report. Exp Ther Med 24(3):574. https://doi.org/10.3892/etm.2022.11511. PMID:35949343;PMCID:PMC9353512
Pomeroy J, Krentz AD, Richardson JG, Berg RL, VanWormer JJ, Haws RM (2021) Bardet-Biedl syndrome: weight patterns and genetics in a rare obesity syndrome. Pediatr Obes. 16(2):e12703. https://doi.org/10.1111/ijpo.12703
Tsyklauri O, Niederlova V, Forsythe E, Prasai A, Drobek A, Kasparek P, Sparks K, Trachtulec Z, Prochazka J, Sedlacek R, Beales P, Huranova M, Stepanek O (2021) Bardet-Biedl Syndrome ciliopathy is linked to altered hematopoiesis and dysregulated self-tolerance. EMBO Rep 22(2):e50785. https://doi.org/10.15252/embr.202050785. Epub 2021 Jan 11. PMID: 33426789; PMCID: PMC7857422
Dervisoglu E, Isgoren S, Kasgari D, Demir H, Yilmaz A (2011) Obesity control and low protein diet preserve or even improve renal functions in Bardet-Biedl syndrome: a report of two cases. Med Sci Monit 17(1):CS12-14. https://doi.org/10.12659/msm.881320. PMID: 21169913; PMCID: PMC3524693
Choo YY, Agarwal P, How CH, Yeleswarapu SP (2019) Developmental delay: identification and management at primary care level. Singapore Med J 60(3):119–123. https://doi.org/10.11622/smedj.2019025. PMID:30997518;PMCID:PMC6441684
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, ACMG Laboratory Quality Assurance Committee (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 17(5):405–24. https://doi.org/10.1038/gim.2015.30. Epub 2015 Mar 5. PMID: 25741868; PMCID: PMC4544753
Álvarez-Satta M, Castro-Sánchez S, Valverde D (2017) Bardet-Biedl syndrome as a chaperonopathy: dissecting the major role of chaperonin-like BBS proteins (BBS6-BBS10-BBS12). Front Mol Biosci 31(4):55. https://doi.org/10.3389/fmolb.2017.00055. PMID:28824921;PMCID:PMC5534436
Dong X, Li Z, Wang D, Xiong Y, Li H, Yang P, Lao L, Man T, Gan Y (2022) Prenatal diagnosis of Bardet-Biedl syndrome due to novel variants in the BBS10 gene in a fetus with multiple anomalies: a case report. Exp Ther Med 24(6):721. https://doi.org/10.3892/etm.2022.11657. PMID:36340607;PMCID:PMC9627110
Ward HH, Brown-Glaberman U, Wang J, Morita Y, Alper SL, Bedrick EJ, Gattone VH 2nd, Deretic D, Wandinger-Ness A (2011) A conserved signal and GTPase complex are required for the ciliary transport of polycystin-1. Mol Biol Cell 22(18):3289–305. https://doi.org/10.1091/mbc.E11-01-0082. Epub 2011 Jul 20. PMID: 21775626; PMCID: PMC3172256
Acknowledgements
Not applicable.
Code availability
Not applicable.
Funding
No funding was received from any source.
Author information
Authors and Affiliations
Contributions
GR—conception of report, manuscript writing and editing along with its substantive revisions, data collection and analysis, preparation of figures and tables. BP—data collection. VDS—design of work and interpretation of data regarding examination findings and investigations of the patient. SS—analysis and revisions in the manuscript. All the authors have accepted responsibility for the entire content of this submitted manuscript and approved submission as well as agreed to be personally accountable for the author’s own contributions. The authors declare that all data were generated in-house and that no paper mill was used.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The case report was submitted and approved by the ethics committee of Topiwala National Medical College and B.Y.L Nair charitable hospital, Mumbai, India. The use of whole exome sequencing was also approved.
Consent for publication
Written informed consent was obtained from the legal guardians of the patient included in the study for publication of this case report and accompanying images.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rekhawar, G.R., Bhavana, M.P., Sawant, V.D. et al. Bardet-Biedl Syndrome: a case report of delayed diagnosis with variable presentation and role of genetic testing in definitive diagnosis. Egypt Pediatric Association Gaz 71, 52 (2023). https://doi.org/10.1186/s43054-023-00196-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43054-023-00196-5