
Abstract
Background
Ankylosing spondylitis (AS) is a type of chronic inflammation that is most prevalent in young adults and is characterized by an inflammatory enthesiopathy that gradually develops toward ossification and ankylosis. If inflammation is left unchecked, it can potentially lead to complications such as secondary amyloidosis, also known as AA amyloidosis, involving the deposition of amyloid serum A protein. Our case presents with a thyroid localization of AA amyloidosis which is secondary to this AS. Such a case has been described in only four cases in the literature. Cardiac localization of AA amyloidosis has been exceptionally described in the literature.
Case presentation
We report the case of a young patient with severe AS complicated by secondary amyloidosis with thyroid, cardiac, and probably renal localization. He was treated with anti-TNF therapy, and his condition improved significantly.
Conclusions
Our case presents several localizations of AA amyloidosis secondary to this AS. Although cardiac involvement is rare in secondary AA amyloidosis, it should always be screened for, even in a cardiacly asymptomatic patient.
Similar content being viewed by others


Background
Secondary amyloidosis (AA) is an important complication of chronic inflammatory diseases. Rheumatologic diseases such as rheumatoid arthritis (RA) and ankylosing spondylitis (AS) are known to be associated with the development of AA amyloidosis. Although AS affects the spine and sacroiliac joints as well as peripheral joints, other extra-articular manifestations are possible and include acute anterior uveitis, aortic insufficiency, apical pulmonary fibrosis, and systemic amyloidosis. AA is a systemic disease characterized by amyloid deposition in many organs. The kidney is the most affected organ, damage to the thyroid gland is possible, but cardiac involvement remains rare during secondary amyloidosis. The present case illustrates a unique presentation of multisystem secondary amyloidosis.
Case presentation
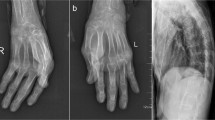
A 33-year-old male patient, on dialysis since the age of 26 for kidney damage of undocumented origin, referred to an internal medicine consultation for the etiological assessment of a large amyloid goiter (Fig. 1) evolving for 3 years with a normal thyroid balance initially. During the interrogation, the patient reported since the age of 16 years inflammatory low back pain associated with buttock pain, heel pain, and enthesopathy, which prompted the patient to self-medicate with nonsteroidal anti-inflammatory drugs if necessary. On clinical examination, the patient presented with kypho-scoliosis, stiffness of the cervical, dorsal, and lumbar spine (Fig. 2) as well as pain on passive and active mobilization of both hips. The rest of the somatic examination was unremarkable, including no cardiovascular symptoms.

Frontal section of a voluminous goiter compressing the laryngeal canal, bumpy contours with heterogeneous density

Kypho-scoliosis with spinal stiffness
Biologically, the assessment was in favor of an inflammatory profile with a C-reactive protein at 87 mg/l, a fibrinogen level at 7 g/l, hypochromic microcytic anemia at 8 g/dl, thrombocytosis at 500,000 / mm3, electrophoresis of serum proteins found hypoalbuminemia at 30 g/l, hyper alpha 1 and alpha 2 globulins. The HLA B27 was positive. Pro-BNP (brain natriuretic peptide) assay was frankly positive at 885 pg/ml.
X-rays were performed (Fig. 3), showing a bamboo spine appearance at the level of the thoraco-lumbar spine, accentuated kyphosis.

Frontal X-ray of the lumbar spine: syndesmophytes, joint pinching, and diffuse bone demineralization
Histology of thyroid tissue (Fig. 4) after thyroidectomy found a thyroid parenchyma where the interstitial tissue is enlarged by an abundant amyloid deposit comprising clusters of mature adipocytes interposed between the thyroid vesicles. A biopsy of the accessory salivary glands was performed (Figs. 5 and 6), which revealed amyloid deposits with positive staining by anti-serum amyloid A (SAA) antibody in immunohistochemistry.

Histological section of a thyroid parenchyma: The interstitial tissue is enlarged by an abundant amyloid deposit comprising clusters of mature adipocytes interposed between the thyroid vesicles (A H&E staining ×200), (B H&E staining ×100)

Histological section of a salivary parenchyma in special Congo red staining marks the deposition in brick red (A Congo red ×200) and characteristic yellow–green birefringence in polarized light (B polarized light ×100)

Histological section of a salivary parenchyma: extracellular areas of anhistic, amorphous material. Amyloid deposits (A H&E staining ×200) with positive staining by anti-SAA antibody in immunohistochemistry (B anti-SAA antibody ×100)
A cardiac assessment was carried out. The electrocardiogram (ECG) (Fig. 7) revealed a sinus rhythm at 75 bpm, with diffuse microvoltage, pseudo-infarct pattern, and negative T-wave from V1 to V3. On transthoracic echocardiography the left ventricle (LV) was normal sized (indexed left ventricular end-diastolic diameter (ILVEDD) = 40 mm/m2), hypertrophied (interventricular septum (IVS) = 11 mm, posterior wall = 16 mm) (Fig. 8), with a normal radial systolic function (left ventricular ejection fraction (LVEF) = 65%), an altered longitudinal function with a mitral annular plane systolic excursion (MAPSE) = 13 mm, and an S’VG = 6 cm/s (Fig. 9), and a global longitudinal systolic strain altered at -17.3% (Fig. 10). We also noted a pseudonormal mitral profile, with biatrial enlargement (left atrium (LA) = 26 cm2, right atrium (RA) = 21 cm2) (Fig. 11). The right ventricular (RV) was not dilated, not hypertrophied, with a normal systolic function. There was a minimal pericardial effusion next to the right cavities (Fig. 12).

The electrocardiogram

Mean LV hypertrophy

Alteration of LV longitudinal function

Alteration of the global longitudinal strain of the LV at − 17.3%

Pseudonormal mitral profile and biatrial dilatation

Low abundance pericardial effusion near the right cavities
The patient received anti-TNF (tumor necrosis factor) treatment, and after 3 months of treatment, there was a significant biological improvement of the inflammatory syndrome.
Discussion
Ankylosing spondylitis is a chronic immune-mediated inflammatory arthritis included in the so-called group of spondyloarthritis (SpA). The axial skeleton and the sacroiliac joints are the primary targets of this disease in males in their third decade of life [1]. Other disabling manifestations, such as inflammatory bowel disease, uveitis, and amyloidosis, may occur in addition to skeletal involvement [2].
Inflammatory amyloidosis, also known as AA amyloidosis (amyloid associated), is a form of generalized amyloidosis, just like AL amyloidosis (immunoglobulinic) and hereditary amyloidosis. In AA amyloidosis, the amyloid protein is the AA protein, which is derived by cleavage from the "serum amyloid associated protein" (SAA), one of the major proteins in the reaction inflammatory [3]. The diagnosis of amyloidosis is based on clinical organ involvement and histological evidence of target organ showing deposition of abnormally folded proteins leading to organ dysfunction. Amyloid deposits are formed from globular, soluble proteins, which undergo misfolding and, subsequently, aggregate into insoluble fibrils, or proteins may also have an intrinsic tendency to form amyloid in the absence of misfolding [4]. The spleen and liver are the first places where AA amyloid deposits appear [5]. Nevertheless, despite significant amyloid infiltration, splenic and hepatic involvement remain asymptomatic for a long time or only lead to mild liver function abnormalities. On the other hand, the deposition of AA amyloid fibrils in the mesangium and in the glomerular capillary walls in the kidney invariably leads to proteinuria, nephrotic syndrome, and the progressive development of renal failure, which are therefore the main features of the presentation of AA amyloidosis [6].
Our patient had undocumented nephropathy which was probably due to renal amyloidosis. Amyloid infiltration of the thyroid gland is possible; however, thyroid amyloid deposits are rarely large enough to result in clinically recognizable goiter; less than 150 cases of amyloid goiter have been described in the literature [7]. Amyloid goiters grow rapidly, while most patients present with a euthyroid state like our patient, cases of hypothyroid, and hyperthyroid states have also been reported [8]. Only four cases of amyloid goiter secondary to ankylosing spondylitis have been described in the literature [7, 9,10,11].
The prevalence of cardiac involvement is unknown in published studies and is likely underestimated [12]; although deposition of AA amyloid can be observed by echocardiography in approximately 10% of patients, restrictive cardiomyopathy progressing to heart failure rarely develops even in stage late in the disease [13]. Diagnosing cardiac involvement in AA amyloidosis can be challenging [14]. Despite an extensive literature search of noninvasive imaging modalities for the diagnosis of cardiac AA amyloidosis, no historical studies or convincing case reports were identified. In one study, clinical heart failure was identified in only one of 374 patients with AA amyloidosis and significant LV hypertrophic on transthoracic echocardiography [15]. In another study of 199 patients with AA amyloidosis, 23 (12%) had heart failure, and cardiac biopsies were performed in 13, all of which showed AA amyloid deposits [16].
The diagnosis of cardiac amyloidosis (CA) is difficult and requires the combination of several clinical data, different imaging, and histological modalities (endomyocardial biopsy) [14]. Patients with CA present with a variable clinical presentation that can range from asymptomatic LV dysfunction detected as part of the assessment of systemic amyloidosis, as is the case of our patient, to refractory heart failure or cardiogenic shock. Usually dyspnea, edema, palpitations, or syncope is the main symptom revealing cardiac involvement [17]. Since the ECG is an easy and accessible examination, it makes it possible to orient toward a diagnosis of cardiac amyloidosis. In a study carried out in 2013 [14], on the various most sensitive and specific electrical aspects in the event of cardiac amyloidosis, we find: low voltage on limb leads, atrial arrhythmia, atrioventricular block, and pseudo-infarct pattern. In cardiac amyloidosis, limb leads with low-voltage and pseudo-infarct pattern were more common.
Echocardiography, which is widely available, has greatly improved the diagnosis of CA after the introduction of strain analysis; despite this, its sensitivity and specificity remain low [14]. In all patients with unexplained LV hypertrophy and clinical suspicion of cardiac amyloidosis, it is recommended to perform full 2D echocardiography, which includes quantitative tissue Doppler and longitudinal strain analysis [18]. Echocardiographic evaluation for signs of amyloidosis such as myocardial wall hypertrophy and biatrial dilation is helpful [17]. The presence of moderate to severe LV thickening (wall thickness ≥ 14 mm) with or without right ventricle (RV) hypertrophy should raise suspicion of CA [14]. Granular appearance scintillation of the myocardial walls can be appreciated [18]. Myocardial contractility can be preserved or even supranormal for a long period. Heart failure occurs at a later stage when left ventricular filling is impaired [14]. Taking the ratio of apical longitudinal strain to basal and median longitudinal strain, a value greater than 1.3 predicts the existence of CA. With the presence of the "cherry-like" apical sparing strain associated with other features such as biventricular hypertrophy, atrial enlargement, a restrictive mitral profile and pericardial effusion. In this context, a more in-depth investigation using other more precise diagnostic modalities such as bone scintigraphy, cardiac magnetic resonance imaging (MRI), should be carried out [19].
Echocardiographic parameters should be combined with electrocardiographic, clinical, biomarker, and other imaging findings to maximize diagnostic accuracy [18]. It should be noted that cardiac biomarkers (troponins and pro-BNP (brain natriuretic peptide)) remain one of the important diagnostic parameters. And the evaluation of these biomarkers is an integral part of the management of patients with cardiac amyloidosis [18].
For this, cardiac MRI remains a powerful examination which makes it possible to differentiate cardiac amyloidosis from hypertrophic cardiomyopathy (HCM) depending on the late gadolinium enhancement (LGE) model.
Despite all the progress, the challenge for effective and rapid diagnosis persists. The only sure diagnosis of cardiac amyloidosis requires an endomyocardial biopsy, which remains the gold standard, as it is virtually 100% accurate. Endomyocardial biopsy is necessary when suspected cardiac amyloidosis is an isolated feature or when the type of cardiac amyloid fibril cannot be identified by other means. In practice, in AA amyloidosis we have other histological evidence. Therefore, the need for endomyocardial biopsy is limited. Cardiac amyloidosis has a poor prognosis, but this differs depending on the type of amyloidosis, stage of heart failure, availability, and response to treatment. The treatment takes several parts: The treatment of heart failure (HF) (beta-blocker, angiotensin-converting enzyme (ACE) inhibitor, or angiotensin II receptor antagonist (ARA II), sodium-glucose cotransporter 2 (SGLT2) inhibitors, or others), which represents the main component, must be used with caution given the risk of complications and harmful drug interactions. The primary goal of treatment for HF is to maintain adequate filling pressures with the balancing peripheral edema and renal failure. Therapies act on the production of amyloid fibril precursor proteins, and new strategies to inhibit the formation of amyloid fibrils or to directly target amyloid deposits. Heart transplantation, although rarely feasible, can be very effective in carefully selected patients. Education, patient involvement, and support are essential for successful management [20]. And finally, the treatment of AA amyloidosis aims to reduce the production of AAS. In addition to etiological treatment with corticosteroids and immunosuppressants, treatment with monoclonal antibodies directed against cytokines, in particular TNF and interleukin-6 (IL-6), is effective in many cases. This type of treatment is targeted not at the level of the amyloid fibrils themselves but rather at the level of the messengers in the acute phase response [21].
Conclusions
AA amyloidosis is rare in ankylosing spondylitis; it is a very serious complication due in particular to the risk of renal damage. Care should be taken in case of cardiac involvement, exceptional in AA amyloidosis, or other unusual organ involvement in AA amyloidosis, such as involvement of the thyroid gland. Our case shows that even when faced with an asymptomatic patient, cardiac involvement must be detected, especially when amyloidosis affects several organs.
Availability of data and materials
Not applicable.
Abbreviations
- AS:
-
Ankylosing spondylitis
- AA:
-
Secondary amyloidosis
- RA:
-
Rheumatoid arthritis
- CA:
-
Cardiac amyloidosis
- BNP:
-
Brain natriuretic peptide
- SAA:
-
Anti-serum amyloid A
- ECG:
-
Electrocardiogram
- LV:
-
Left ventricle
- ILVEDD:
-
Indexed left ventricular end-diastolic diameter
- IVS:
-
Interventricular septum
- LVEF:
-
Left ventricular ejection fraction
- MAPSE:
-
Mitral annular plane systolic excursion
- LA:
-
Left atrium
- RA:
-
Right atrium
- RV:
-
Right ventricular
- TNF:
-
Tumor Necrosis Factor
- MRI:
-
Magnetic resonance imaging
- HCM:
-
Hypertrophic cardiomyopathy
- LGE:
-
Late gadolinium enhancement
- HF:
-
Heart failure
- ACE:
-
Angiotensin-converting enzyme
- ARA II:
-
Angiotensin II receptor antagonist
- SGLT2:
-
Sodium-glucose cotransporter 2
- IL-6:
-
Interleukin-6
References
Garcia-Montoya L, Gul H, Emery P (2018) Recent advances in ankylosing spondylitis: understanding the disease and management [version 1; referees: 2 approved]. F1000Research , 7:1512. https://doi.org/10.12688/f1000research.14956.1
Sang Youn Jung, Min-Chan Park, Yong-Beom Park, and Soo-Kon Lee. Serum Amyloid A as a Useful Indicator of Disease Activity in Patients with Ankylosing Spondylitis. Yonsei Med J. 2007 Apr 30; 48(2): 218–224.
Stojanovic KS et al (2017). Amylose AA Néphrol ther. https://doi.org/10.1016/j.nephro.2017.03.001
Picken MM (2020) The pathology of amyloidosis in classification: a review. Acta Haematol 143:322–334. https://doi.org/10.1159/000506696
Hawkins PN (2002) Serum amyloid P component scintigraphy for diagnosis and monitoring amyloidosis. Curr Opin Nephrol Hypertens 11:649–655
Hazenberg BP, van Rijswijk MH (1994) Clinical and therapeutic aspects of AA amyloidosis, Bailliere’s. Clin Rheumatol 8:661–690
Cohan P, Hirschowitz S, Yu Rao J, Tanavoli S, Van Herle AJ (2000) Amyloid goiter in a case of systemic amyloidosis secondary to ankylosing spondylitis. J Endocrinol Invest 23:762–764
Amado JA, Palacios S, Manzanos J (1982) Fast growing goitre as the first clinical manifestation of systemic amyloidosis. Postgrad Med J 58:171–172
Hocaoglu E, Aydemir E, Ates C, Saridas FM, Cander S, Gul OO, et al. A rare case of symptomatic amyloid goiter diagnosed by tru-cut biopsy. Endocrine Abstracts (2022) 81 EP969.
Elaouni, Soukaina, Ahmed J, Fouad Z, Zakia B, Omar E, et al. “Rapid Growing Amyloid Goiter Mimicking a Malignant Thyroid Tumor in 24-Year-Old-Male, Secondary Amyloidosis and End-Stage kidney Failure.” Clin Med Case Rep 5 (2021): 177
Selim Bakan, Sedat Giray Kandemirli, Serkan Akbas, Mehmet Cingoz, Burcu Ozcan Guzelbey, Fatih Kantarci, Canan Akman. Amyloid Goiter: A Diagnosis to Consider in Diffuse Fatty Infiltration of the Thyroid. J Ultrasound Med. 2017 May;36(5):1045–1049. doi: https://doi.org/10.7863/ultra.16.04037.
Sanchez AC, Murphy R, Rao S, Martinez F, Bryant S, Chaudhuri D (2021) A Case Report of Cardiac Amyloidosis Highlighting the Importance of Strain Analysis. Case Rep Cardiol 12(2021):1–5. https://doi.org/10.1155/2021/5673364
Obici L, Perfetti V, Palladini G, Moratti R, Merlini G (2005) Clinical aspects of systemic amyloid diseases. Biochem Biophys Acta 1753:11–22
Li B, Ahluwalia M, Narula N, Moreira AL, Swistel DG, Massera D, Sherrid MV (2020) Cardiac AA Amyloidosis in a Patient with Obstructive Hypertrophic Cardiomyopathy. Cardiovasc Pathol. https://doi.org/10.1016/j.carpath.2020.107218
Hamer JP, Janssen S, van Rijswijk MH, Lie KI. Amyloid cardiomyopathy in systemic nonhereditary amyloidosis. Clinical, echocardiographic and electrocardiographic findings in 30 patients with AA and 24 patients with AL amyloidosis. Eur Heart J. 1992;13:623–7.
Okuda Y, Yamada T, Ueda M, Ando Y (2018) First Nationwide Survey of 199 Patients with Amyloid A Amyloidosis in Japan. Intern Med 57:3351–3355
Kittleson MM, Maurer MS, Ambardekar AV, Bullock-Palmer RP, Chang PP, Eisen HJ, et al. Cardiac Amyloidosis: Evolving Diagnosis and Management: A Scientific Statement From the American Heart Association. Circulation, Volume 142, Issue 1, 7 July 2020; Pages e7-e22. https://www.ahajournals.org/doi/https://doi.org/10.1161/CIR.0000000000000792
Sharmila Dorbala, MD and al. SNC/AHA/ASE/EANM/HFSA/ISA/SCMR/ SNMMI Expert Consensus Recommendations for Multimodality Imaging in Cardiac Amyloidosis. Part 1 of 2—Evidence Base and Standardized Methods of Imaging. Circ Cardiovasc Imaging. 2021;14:e000029. DOI: https://doi.org/10.1161/HCI.0000000000000029.
Anatoli Kiotsekoglou MD, PhD and al. Echocardiographic diagnosis of cardiac amyloidosis: Does the masquerader require only a "cherry on top"? Echocardiography. 2020;37:1713–1715. DOI: https://doi.org/10.1111/echo.14952.
Sanjay M. Banypersad, MRCP, and al. Updates in Cardiac Amyloidosis: A Review. J Am Heart Assoc. 2012;1:e000364 doi: https://doi.org/10.1161/JAHA.111.000364
Westermark GT, Fandrich M, Westermark P (2015) AA Amyloidosis: Pathogenesis and Targeted Therapy. Annu Rev Pathol Mech Dis 10:321–344
Acknowledgements
None.
Funding
None.
Author information
Authors and Affiliations
Contributions
Dr. BL conducted the literature review, designed, and wrote the manuscript. Dr EK, MM, EH revised the literature review. Dr EY, HM, AS, BMG, DA, and HR wrote the cardiological part. Dr AM, MA, RM, BGN, and KM provided us with the anatomo-pathological images as well as their interpretation. All authors have read and accepted the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approval was exempted by the Ethical Committee at Ibn Roch University Hospital for reporting this case.
Consent for publication
Written informed consent was obtained from the patient for the publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal on request.
Competing interests
The authors report no declarations of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Barakat, L., Echchilali, K., Moudatir, M. et al. A rare clinical case of systemic AA amyloidosis with cardiac involvement complicating ankylosing spondylitis: a case report. Egypt Heart J 76, 40 (2024). https://doi.org/10.1186/s43044-024-00471-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43044-024-00471-9