
Abstract
Background
Hydrocephalus is one of the most common pathophysiological disabilities with a high mortality rate, which occurs both congenitally and acquired. It is estimated that genetic components are the etiology for up to 40% of hydrocephalus cases; however, causal mutations identified until now could only explain approximately 20% of congenital hydrocephalus (CH) patients, and most potential hydrocephalus-associated genes have yet to be determined. This study sought to find causal variations in a consanguineous family with four affected children diagnosed with hydrocephalus.
Material and methods
In this study, we evaluated twenty-five members of an extended family consisting of a nuclear family with four affected children resulting from a consanguineous couple and eighteen of their relatives, including one hydrocephalus case. The mother of this family was experiencing her 15th week of pregnancy, and cytogenetic evaluation was performed using amniocentesis to identify fetal chromosomal abnormalities. We conducted whole-exome sequencing (WES) on the genomic DNA of the proband to detect the CH-causing variants, followed by confirmation and segregation analysis of the detected variant in the proband, fetus, and family members through Sanger sequencing.
Results
Following the bioinformatic analysis and data filtering, we found a homozygous variant [NM_001243766.2:c.74G>A:p.W25X] within the protein O-mannose beta-1,2-N-acetylglucosaminyltransferase 1 (POMGNT1) gene confirmed by Sanger sequencing in the proband and segregated with the hydrocephalus in the family. The variant was described as pathogenic and regarded as a nonsense-mediated mRNA decay (NMD) due to the premature stop codon, which results in a truncated protein.
Conclusion
The results of the current study broadened the mutational gene spectrum of CH and our knowledge of the hydrocephalus etiology by introducing a novel homozygous variant within the POMGNT1 gene, which had never been previously reported solitary in these patients.
Similar content being viewed by others

Introduction
Hydrocephalus is a complicated and poorly understood condition affecting any age, from fetal development to adulthood [1]. Hydrocephalus is approximately 4.65 per 10,000 live births and is seen in about 6.4 million individuals worldwide [2]. This pathophysiological condition may be congenital or develop due to trauma, infection, venous occlusion, tumors, or cerebrospinal fluid (CSF) buildup in the ventricular space due to under-absorption, over-production, or a blockage of CSF flow [3]. According to epidemiological studies and reports of familial congenital hydrocephalus (CH), genetic components are estimated to be the etiology for up to 40% of hydrocephalus cases [2, 4]. Although over 100 genes have been described to be mutated in syndromic hydrocephalus cases, few causal mutations have been identified, which only could explain approximately 20% of CH patients [5, 6]. Among these candidate genes, eight mutated genes follow the Mendelian inheritance pattern and are related to non-syndromic hydrocephalus. The X-linked type of the non-syndromic hydrocephalus includes L1CAM and AP1S2 mutations and autosomal recessive hydrocephalus associated with CCDC88C, MPDZ, TRIM71, SMARCC1, PTCH1, and SHH mutations [7, 8].
Ultrasound examination is an essential first-line screening approach to help us identify CH in the fetus. However, due to the genetic heterogeneity and non-specific phenotype of CH in the fetal period, ultrasound alone is not enough to make a prenatal diagnosis and distinguish the causes of CH [9]. Next-generation sequencing (NGS) clinical application allows specific early interventions for the identified CH after birth. Besides, it provides optimal birth management because the molecular characterization of CH has fundamental implications in the clinical setting [10,11,12]. Whole-exome sequencing (WES) could improve the prenatal diagnostic yield and expand our knowledge concerning the etiologies of CH by sequencing all DNA coding parts [11, 13]. In this study, we evaluated twenty-five members of an extended family consisting of a nuclear family with four affected children and eighteen of their relatives that also included one hydrocephalus case. We carried out whole-exome sequencing in an infant of the nuclear family (proband) to detect the CH-causing variants, followed by confirmation of the identified variant in the proband and segregation analysis in his family members through Sanger sequencing. We finally ended up with a novel pathogenic nonsense variant [NM_001243766.2:c.74G>A:p.W25X] within the POMGNT1 gene at the homozygous state that was confirmed and segregated in the proband’s family members. Clinical genetic evidence indicates that all POMGNT1 variants listed in genetic databases are linked to the various phenotypes of congenital muscular dystrophy dystroglycanopathy (MDDG), such as type A-3 (congenital with brain and eye anomalies), type B-3 (congenital with impaired intellectual development), type C-3 (limb–girdle), and retinitis pigmentosa-76 (RP76) [14, 15]. In addition to the abnormalities mentioned above, compound heterozygous variants of POMGNT1, alongside other variations, have also been reported in congenital nervous system disorders and a handful of hydrocephalus cases [16]. However, other than the above-mentioned abnormalities, these variants have not been described as pathogenic in other neuropathogenic conditions. This study is the first report of this exonic POMGNT1 variant solitary in CH patients, expanding the variant spectrum of the POMGNT1 gene and highlighting their potential in the CH pathogenicity.
Material and methods
Ethical considerations and informed consent
The Islamic Azad University, Tehran Medical Sciences Branch approved the study, and all the experiments were conducted following the standard ethical protocols of the institutional and national research committee. Informed consent was obtained from all participants in this investigation and parents as the proband’s legal guardians to publish any potentially identifiable data in this article.
Clinical characteristics of the proband and patients
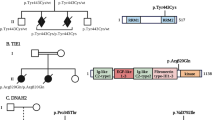
A consanguineous couple with four affected children for evaluation of familial CH was referred to our prenatal diagnosis department, Noorgene Genetic & Clinical Laboratory, Ahvaz, Iran (ClinVar-Organization: https://www.ncbi.nlm.nih.gov/clinvar/submitters/508856/). Ventriculomegaly and functional deficits were diagnosed and confirmed in all four children (aged from 4 days to 15 years at the evaluation time). Stuttering and dysarthria were clinically recognized in this family’s first and second children, and seizure was also present in the third child, as in the first (Table 1). The parents were healthy, and no further cases were known in the extended family except for a male paternal cousin who suffered from hydrocephalus and recurring seizures. The asymptomatic parents were the first cousin, and the 30-year-old woman was in her fifth pregnancy at 15 weeks of gestation (Fig. 1). The proband was an affected 4-day-old boy who shared a phenotype of CH and multiple congenital anomalies. The proband’s head circumference at birth ranged from 324 to 331 mm, and he was born through an uneventful delivery at 39th weeks of gestation with 2602 ± 100 gr birth weight (Table 2). A standard ultrasound proband screening at the 22nd week of pregnancy revealed moderate-to-severe hydrocephaly and dilated bilateral lateral ventricles with 66 mm biparietal diameter (BPD) and 245 mm abdominal circumference (AC). Ultrasound of the proband at 27 weeks of gestation exhibited dilatation of the cerebral posterior horns bilaterally (12 mm [left], 29 mm [right]) and agenesis of the corpus callosum. The last screening at 34 weeks of gestational age revealed 90 mm BPD, 324 mm head circumference, 304 mm AC, and the parenchymal atrophy of lateral and third ventricles was measured at 13 and 33 mm, respectively. The couple wanted to find out the etiology of their children’s consecutive histories of hydrocephalus. We offered a whole-exome sequence for this couple. The parents also requested genetic testing for their fetus in the present pregnancy.

Pedigree of the nuclear family with four affected siblings and extended family members. On the pedigree, the genotypes of the consanguineous parents and their afflicted offspring for the c.74 G>A variation are displayed. The afflicted proband is denoted by the arrow. Abbreviations: Wks: Weeks; A: Adenine; G: Guanine
Prenatal cytogenetic analysis
The first-trimester ultrasound screening revealed no sonographic abnormalities, and the fetus had normal heartbeats with a regular nuchal translucency (NT) measurement (1.5 mm). Following genetic consultation at 15 weeks of gestation, we performed an amniocentesis procedure using a freehand technique with an ultrasound guide and a 20-gauge needle. A 20 ml of amniotic fluid was collected and divided into two parts; 12 ml of amniotic fluid was transferred to the cytogenetic section of our laboratory to place in culture, and the rest (8 ml) was used for fetal molecular testing. Following centrifugation, amniotic fluid was subjected to in situ Amniomax-II Complete Medium containing Fetal Bovine Serum, Gentamicin, and L-Glutamine for six days at 37 °C (5% CO2). Before the cells were harvested, semi-confluent cells were treated with colchicine (final concentration of 75 ng/mL) and incubated at 37 °C for about 20 min to arrest the cells in metaphase. After removing the mitogen-containing media, the cells were thoroughly rinsed with pre-warmed PBS and treated with trypsin for more detachment. In continuation, the cells were treated with 2 mL of pre-warmed 0.075 mM hypotonic KCl solution dropwise. The chromosomes were then fixed with the methanol acetic acid fixative (with the 3:1 ratio of methanol and glacial acetic acid) and washed two times by repeated aspiration, fresh fixative resuspension, and centrifugation. Finally, metaphase spreads were prepared by dropwise application of cell suspension onto clean glass slides. After air-dried for 24 h, the glass slides were incubated with 0.025% trypsin for 1 min and stained with 10% Giemsa solution for about 5 min. The G-banded chromosomes were prepared as described elsewhere [17, 18]. Finally, the metaphase spreads were photographed using Zeiss Imazer.Z2 microscope equipped with GenASIs Case Data Manager system (version 7.2.2.40970, ASI, Carlsbad, CA, USA).
Whole-exome sequencing
Firstly, genomic DNA was extracted based on the salting-out standard protocol from the whole blood of all nuclear family members, proband, and fetus-amniotic fluid cells. Proband genomic DNA was used for WES. Exon capture and enrichment were performed on genomic DNA using the Agilent SureSelectXT Human All Exon V8 (Agilent, Santa Clara, CA) and whole coding regions sequenced on an Illumina HiSeq2000 sequencer (Illumina, Inc., San Diego, CA, USA) with an average depth of 100x. The raw whole-exome sequencing data were examined using a cori7 central processing unit and the Linux operating system (Ubuntu 20.04). Various in silico prediction tools and bioinformatics resources were then used to give additional support for the found variations according to the following steps:
-
Quality control (QC) of the read lengths was surveyed using the FastQC tool (version 0.11.9) based on GC content ~ 50% and a Phred value = 20. The Trimmomatic software (version 0.36) removed low-quality and adapter-contaminated reads.
-
Sequence reads precisely were mapped to the human reference genome (GRCh37/hg19) with Burrows–Wheeler Aligner (BWA) aligning tool (Version 2.4.0), and outputs were generated in Sequence Alignment/Map (SAM) file format. Conversion of the SAM file to the Binary Alignment Map (BAM) file was conducted through the Picard tool.
-
The local realignment of insertion/deletions (indels), base quality score recalibration (BQSSR), single-nucleotide variants (SNVs)/indels calling, and data compression was performed and filed in variant call format (VCF) using a combination of Genome analysis toolkit Haplotypecaller (GATK HC) and VarScan mpileup-2snp protocol.
-
The outpouring VCF file was annotated employing Gene Ontology Reference Genome Project, multiple association network integration algorithm (GeneMANIA), protein analysis through evolutionary relationships (PANTHER), and Ensembl variant effect predictor (Ensembl VEP).
The annotated variants were filtered using the R programming command-line software by the following steps:
-
The minor allele frequencies (MAFs) of all known variants were annotated according to the dbSNP, 1000 Genome Project, Exome Sequencing Project (ESP), Genome Aggregation Database (gnomAD), Iranome (https://www.iranome.ir/), and Exome Aggregation Consortium (ExAC) databases.
-
Synonymous, deep intronic, and less conserved variants were excluded using Combined Annotation Dependent Depletion (CADD) PHRE and GERP scores.
-
The potential or known genes involved in the CH pathogenesis were distinguished from genes not relating to the CH based on the provided clinical/paraclinical findings through the phenotype-based prioritization (Phenolyzer) tool and the human phenotype ontology (HPO) (https://hpo.jax.org/).
-
The remaining variants were evaluated and classified considering the American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) rules [19].
-
Finally, the depth of reads for each variant was scrutinized using the Integrative Genomics Viewer (IGV) software.
Variant confirmation and family segregation
Genomic DNA was extracted based on the salting-out standard protocol from the whole blood of the proband and all family members and from the fetus’s amniotic fluid cells. The primer sequences covering the WES-identified variant in the proband were designed through Primer3 web-based software (https://bioinfo.ut.ee/primer3-0.4.0/). The OligoAnalyzer Tool was employed to examine the features and quality of the Forward-primer (POMGNT1-EX2-F-248: CTG GTC CCT TCA GAT TCC TGA AG) and Reverse-primer (POMGNT1-EX2-R-248: TGA TTT AGT GGG GAG GAA GCT G). Subsequently, Sanger sequencing was carried out following the PCR amplification for the region harboring the WES-identified variant to validate the potential causal variant in the proband, his family members, and the fetus.
The PCR reaction was performed using the FlexCycler Thermocycler. PCR reaction for the identified variant was optimized to amplify the desired region as follows: 1 μl of DNA (concentration: 50 ng/μL) was used as a template in 12.5 μl reaction mixture (containing dNTPs, 1× PCR buffer, MgCl2, and Taq polymerase), 0.5 μl of each primer (concentration: 20 μM), and 10 μl of distilled water (the total volume of the reaction solution: 25 μL). The PCR procedure for the desired region included the initial denaturation at 94 °C for 5 min, followed by 35 cycles of the next steps: denaturation at 94 °C for 1 min, annealing at 61 °C for 45 s, elongation at 72 °C for 1 min, and final elongation at 72 °C for 4 min. After the PCR reactions, 6 μl of PCR product was loaded on 2% Agarose gel in × 1 Tris–borate–EDTA buffer (PAYA PAZHOOHESH, Iran) stained with DNA-safe stain. Electrophoresis was run at 115 voltages for 50 min. The gel was then inserted into a gel documentation system (under UV light) and analyzed by UV-pro software. The amplicons generated in PCR reactions were sequenced by the Sanger method (using the inclined primers and PCR conditions) and analyzed on an ABI3130XL Genetic Analyzer (USA) to validate PCR amplification and WES results. Eventually, Sanger sequencing files were assembled and aligned through the Codoncode aligner software (version 9.0.1).
Results
Prenatal cytogenetic results
Following the culture of the amniotic fluid and preparation of the G-banded chromosomes, 20 metaphase spreads were assessed. According to the findings, there were no cytogenetic abnormalities, and the chromosomes had a normal appearance with no cleavage or aneuploidy. Furthermore, there were no signs of known chromosomal aberrations associated with hydrocephalus or other congenital anomalies. The 46, XY karyotype revealed that the fetus was male. Figure 2 presents the karyogram of G-banded chromosomes.

Chromosomal analysis of amniotic cells. According to the results, no cytogenetic abnormalities were present, and the chromosomes seemed normal, without any cleavage or aneuploidy. It should be noted that the lack of chromosomal aberration does not rule out the presence of gene abnormalities. Even in cases where the karyotype appears to be normal, low-level mosaicism and other subtle genetic alterations may exist outside the scope of the preparation
Exome sequencing results and segregation analysis
After performing the filtering steps, we ended up with a novel pathogenic homozygous variant [NM_001243766.2:c.74G>A:p.W25X] within the POMGNT1 gene, which was absent from public databases and not documented in the Genome Aggregation Database (gnomAD; https://gnomad.broadinstitute.org), National Center for Biotechnology Information Single Nucleotide Polymorphism Database (dbSNP; www.ncbi.nlm.nih.gov/SNP), Human Gene Mutation Database (HGMD; www.hgmd.cf.ac.uk/ac/), 1000 Genomes Project database (http://www.internationalgenome.org/1000-genomes-browsers/), Ensembl (http://grch37.ensembl.org/index.html), ESP (https://evs.gs.washington.edu), ExAC (https://exac.broadinstitute.org), and Iranome (http://www.iranome.ir) (Table 3). Segregation analysis confirmed this variant in the proband’s family members (Fig. 3). Sanger sequencing demonstrated the homozygous state of the c.74G > A substitution in the proband (TAG/TAG) and his affected siblings (TAG/TAG), as well as the heterozygous state in the parents (TGG/TAG). In contrast to other family members, Sanger sequencing of the fetus revealed no changes in the c.74G, and he had a homozygous state for the wild type (G) in this position (TGG/TGG). In conclusion, the POMGNT1 variant was segregated with hydrocephalus in this family, which could be considered the proband disease-causing.

Nuclear family’s sequencing chromatographs and agarose gel electrophoresis. Sanger sequencing demonstrated that both parents were carriers of the c.74 G>A variant in the POMGNT1 gene and confirmed the homozygous G>A alteration in the proband. Abbreviations: A: Adenine; G: Guanine; C: Cytosine; T: Thymine; L: Ladder; P: Proband; F: Father; M: Mother; and NTC: No-Template Control
Variant analysis
Based on prediction tools including Mutation Taster, CADD, and DANN, the nonsense c.74G>A:p.W25X variant was predicted to be disease-causing, deleterious, and higher probably damaging, respectively (Table 4). It was classified as a pathogenic variant (class 1) according to the ACMG recommendations based on the following rules: PVS1, PM2, and PP3 [19]. Notably, the PP4 criteria can also be applied, as our patient’s clinical symptoms were compatible with the previously reported patients caused by pathogenic variants in the POMGNT1 gene. The variant’s pathogenicity was predicted using the Mutation Taster simulations (https://www.genecascade.org/MutationTaster2021/) as “Deleterious” and regarded as a nonsense-mediated mRNA decay (NMD) due to premature termination and resulting truncated protein (Class “A,” with a score of 1.00 out of 1.00). The Taster simulations predicted that the amino acid sequence would be changed, and protein features (might be) affected because of truncated protein. In addition, the Ensemble Variation Predictor (VEP) analysis, which uses a combination of algorithms such as SIFT and PolyPhen-2, described the pathogenicity of this variant as “Damaging” (http://www.ensembl.org/). FATHMM-MKL and PROVEAN also predicted the c.74G>A:p.Trp25Ter (Termination) alteration as a “Damaging” variant. We also applied pathogenic mutation prediction (PMut) analysis for predicting the pathology score of our variant (https://mmb.irbbarcelona.org/PMut/), which showed a total of 19 variations for Tryptophan at p.25 of POMGNT1 protein. According to the analysis outputs, all substitutions at p.25 were inferred pathogenically and predicted to result in a “Disease” phenotype with an average score of 0.76 out of 1.00 (76%). Besides, a CADD-PHRED score of 36 indicates this variant is among the top 0.1% of most deleterious substitutions possible, and a DANN score of 1 further predicts this variant to be damaging.
On the other hand, evolutionary conservation scanning was carried out through the UCSC genome browser (https://genome.ucsc.edu/), indicating that Tryptophan (W) at position-25 of the POMGNT1 protein sequence is moderately conserved among different species and matching alignments as follows, all identical in Ptroglodytes, as well as partly conserved in Mmulatta, Fcatus, Mmusculus, Ggallus, Trubripes, and Xtropicalis. These findings are substantiated by the outputs derived from the PhastCons100way_vertebrate (1.00), PhastCons30way_mammalian (1.00), PhyloP100way_vertebrate (4.69), PhyloP30way_mammalian (1.026), and SiPhy_29way_logOdds (14.56) datasets (Table 4). Conservation analysis for the p.25W was also performed by the ConSurf server (https://consurf.tau.ac.il/), which described p.25W as a buried residue based on the neural network algorithm. Furthermore, the I-Mutant2.0 server (https://folding.biofold.org/), which computes the ΔΔG values of variations (DG “new protein”—DG “wild type”), determined that the p.Trp25Ter alteration reduces the stability of the POMGNT1 protein (ΔΔG = − 2.00 kcal/mol, ΔΔG < 0: decrease stability).
The c.74G>A variant results in a premature stop codon which can lead to nonsense-mediated mRNA decay (NMD) or a truncated protein, indicating a loss of function. Introducing a premature stop codon leads to a truncated protein missing the remainder of the typical protein sequence. This can impact protein stability and function in several ways. The truncated protein will miss critical functional domains in the C-terminus. Interactions with binding partners may also be reduced or abolished. Furthermore, the truncation can disrupt correct protein folding, impairing protein stability. Nonsense-mediated decay of the mRNA may also reduce the overall expression levels of the protein. Structural modeling showed this variant localized to the stem region, truncating the entire catalytic domain, which could be associated with a complete loss of enzyme activity in functional assays (Fig. 4).

The POMGNT1 protein’s 3D structure and a schematic depiction of its subunits. The canonical isoform of the POMGNT1 protein (Q8WZA1-1) comprises the cytoplasmic tail, transmembrane domain (Signal-anchor for type II membrane protein), catalytic and carbohydrate-binding stem domains. Tryptophan is topologically positioned at p.25 of the POMGNT1 protein and is part of the cytoplasmic tail (Met1 to Arg37)
Discussion
Hydrocephalus is a central nervous system malformation that can occur at any age and result in severe pathological outcomes [20]. The most common findings in these patients are genetic factors, which can be seen in 40% of cases of hydrocephalus [21]. The etiology of CH in the prenatal or postnatal period cannot be identified and distinguished solely by ultrasound due to genetic heterogeneity and non-specific CH phenotypes [22, 23]. Since only 20% of patients possess known mutated genes, indicating that many pathogenic factors hidden in genes have gone undiscovered [6, 14]. The development of molecular testing methods has made it possible to use WES as a quick and accurate method for identifying the causal variants of congenital disorders [11]. In this research, we looked at an extended family group that included a nuclear family with four affected children from a consanguineous couple and eighteen of their relatives. We applied WES on the proband’s genome, followed by segregation analysis using Sanger sequencing in the fetus and family members to identify the variants that cause CH. The identified variant was searched against several major public genomic databases and resources to determine its novelty. These included the gnomAD, which aggregates data from various large-scale genome sequencing projects and was queried (https://gnomad.broadinstitute.org). The dbSNP, a repository for variations including single-nucleotide polymorphisms and multi-nucleotide polymorphisms for human and model organism genomes (www.ncbi.nlm.nih.gov/SNP), and the HGMD, an archive of germline mutations and disease-causing variants maintained by Cardiff University, were also checked (www.hgmd.cf.ac.uk/ac/). In addition, the 1000 Genomes Project database containing common human genetic variations, as well as the publicly accessible genome browser and variation database hosted by EMBL-EBI called Ensembl (http://grch37.ensembl.org/index.html), were evaluated. Further resources consulted included the NHLBI ESP database (https://evs.gs.washington.edu) and the ExAC database (https://exac.broadinstitute.org). Finally, the Iranome database cataloging genetic variations identified in the Iranian population was searched (http://www.iranome.ir). The pathogenic POMGNT1 variant identified in the proband had not previously been reported as a solitary variant in CH patients. This variation was found in all affected children in the homozygous state and the parents in the heterozygous state. In contrast, this variant was not identified in any of the fetus’ haplotypes, and he was completely healthy, unlike other family members.
POMGNT1 gene (chro.1p34.1, ENSG00000085998, OMIM:606822) encodes an integrin membrane protein, and O-linked-mannose beta-1,2-N-acetylglucosaminyltransferase 1 (type II transmembrane protein) exclusively catalyzes the transfer of N-acetylglucosamine residues to alpha-O-linked terminal mannoses, which considered as an essential for the proper functioning of the α-dystroglycan (α-DG) [24]. The α-DG is a subunit of the dystrophin-associated protein complex (DAPC) that, in the glycosylated state, plays a vital function in maintaining the integrity of muscles, brain cell function, and neurological development [25]. Genetic variants affecting the POMGNT1 gene and transcripts regions can decrease the vital function of the POMGNT1 protein and cause hypo-glycosylation of the α-DG, followed by severe neuromuscular impairments [26]. Numerous SNPs and more than 12 transcripts have been presented for the POMGNT1 gene. Various forms of congenital muscular dystrophy dystroglycanopathy (MDDG) associated with POMGNT1 variants, including a severe congenital type of muscular dystrophy with brain and eye anomalies which were known earlier as Walker–Warburg syndrome (WWS) or muscle–eye–brain disease (MEB; OMIM:253280), a less severe congenital form with mental retardation (OMIM:613151), and a milder limb–girdle form with or without normal intellect (OMIM:613157) also known as LGMDR15 and LGMD2O, have been reported singly or in combination with other genetic or acquired syndromes/abnormalities [15]. POMGNT1 gene product consists of the cytoplasmic tail (1–37 residues), transmembrane domain (38–58 residues), and the remaining lumenal region, including carbohydrate-binding stem domain and catalytic domain, localized in the Golgi apparatus (Fig. 4) [27]. However, different protein isoforms have been identified as POMGNT1 gene products due to the existence of distinct spliced transcript variants. The canonical isoform of the POMGNT1 protein includes 646 amino acids. The POMGNT1 transcript variant-2 (ENST00000371992.1, NM_001243766.2) uses an alternate splice site in the CDS, compared to variant-1 (ENST00000371984.8, NM_017739.4) and encodes the isoform 2 of POMGNT1 protein (Q8WZA1-2, ENSP00000361060.1, NP_001230695.2). This isoform is longer (748-amino acids) and has different 5' and 3' UTRs, as well as C-terminus compared to the isoform-1 (Q8WZA1-1, ENSP00000361052.3, NP_060209.4) [28]. There are 23 exonic regions in the POMGNT1 transcript variant-2 (the first coding region begins from the 2nd exon), annotated with 12 domains, features, and 9617 variant alleles (https://www.ensembl.org/). Here, we identified a homozygous nonsense variant within the exon 2 of the POMGNT1 gene in a consanguineous pedigree with multiple members affected by CH. This variation leads to the TGG>TAG replacement at c.74 in the mutated-CDS (mut-CDS). Considering that the stop codon is located at the c.2247 in wild-type-CDS (wt-CDS), this alteration causes a premature stop codon leading to NMD, stalling translation after tryptophan encoding, and a truncated protein product lacking enzymatic function. Topologically, the protein area affected by this nonsense variant belongs to the cytoplasmic tail (Met1 to Arg37), which NMD effects lead to the placement of a premature stop codon at p.25 in the mu-protein sequence instead of p.749 in the wt-protein sequence (https://www.uniprot.org/). In silico analyses overwhelmingly predict this variant to be deleterious, and ACMG criteria classify that as pathogenic. The truncated protein will likely have reduced stability and impaired interactions, ultimately resulting in loss of regular activity. According to the role of the POMGNT1 enzyme in transferring N-acetylglucosamine residues to alpha-O-linked terminal mannose molecules (glycosylation) and subsequently the stability of vital proteins, the variant c.74G>A could lead to a premature stop codon (p.W25X), which causes the POMGNT1 enzyme to be truncated after only 25 amino acids. This truncated enzyme lacks the glycosyltransferase functional domain, which could not efficiently glycosylate specific proteins such as α-DG. The extracellular α-DG is an essential glycoprotein that connects the extracellular matrix (ECM) to the intracellular cytoskeleton in various tissues, including the brain. The α-DG links brain basement membranes (basal laminae) surrounding ventricles to the astroglial endfeet via laminin binding. Impaired cell–matrix adhesion in the brain is believed to disrupt basal laminae structures that line fluid-filled cavities (ventricles) in the brain. This physical disturbance compromises the basement membrane’s ability to act as a barrier and maintain fluid equilibrium. Over time, dysfunction and disruption of these ventricle-lining basal laminae contribute to abnormal accumulation of CSF within the ventricles, leading to ventricles enlarging and compressing surrounding brain structures, followed by hydrocephalus phenotypes and neurological manifestations. However, further in vitro functional studies are needed to conclusively determine the molecular consequences and correlate with the observed patient phenotype. Biallelic pathogenic variants in POMGNT1 have previously been associated with muscle–eye–brain disease and Walker–Warburg syndrome, characterized by congenital muscular dystrophy and structural brain, as well as eye anomalies. Hydrocephalus has occasionally been reported as part of these syndromes but not as an isolated manifestation. The critical difference between the phenotype associated with this novel variant and other POMGNT1 variants lies in the specificity and severity of the presentation. Previous research has shown that POMGNT1 variants near the 5'-terminus are associated with more severe neurodevelopmental malformations than variants near the 3'-terminus. The novel variant identified in this study is characterized by its association solely with hydrocephalus, without the broader range of symptoms typically associated with MDDG types A-3, B-3, C-3, or RP76. The isolated hydrocephalus presentation in patients with the novel POMGNT1 variant can be attributed to the specific nature of the mutation and its impact on the protein’s function. The premature stop codon introduced by the variant probably leads to a loss of function in a critical region of the POMGNT1 protein that is specifically important for the normal development of the brain’s ventricular system. This truncated protein may disrupt the normal glycosylation of α-DG, crucial for maintaining the brain’s structural integrity and preventing cerebrospinal fluid accumulation that characterizes hydrocephalus. Our study is the first to link an isolated POMGNT1 variant to CH without muscular dystrophy. These findings expand the mutation spectrum associated with hydrocephalus and further highlight the importance of POMGNT1 in CNS development.
According to clinical genetic evidence, all POMGNT1 variants reported in genetic databases are associated with MDDG’s different phenotypes, including type A-3 (congenital with brain and eye anomalies), type B-3 (congenital with impaired intellectual development), type C-3 (limb–girdle), and Retinitis pigmentosa phenotype (https://www.omim.org/entry/606822#allelicVariants). A recent Dongmei Fei et al. study reported a compound heterozygous pathogenic variant [c.919C>T:p.Arg307Ter and c.1100del:p.Phe369fs] in FKTN gene (NM_006731.2) along with two compound heterozygous likely pathogenic variants including [c.1449_14-50insACAACG and c.1490G>C:p.Arg497Pro] in the POMGNT1 gene (NM_017739.3), and [c.2690+1G>A and c.1447C>T:p.-Arg483Cys] within the LAMB1 gene (NM_002291.2) in three fetuses with hydrocephalus [16]. They inferred that their reported POMGNT1 variants near the 30 terminus were characterized only by hydrocephalus in fetuses, consistent with previous research results, which had shown more severe neurodevelopmental malformations for the POMGNT1 variants near the 50-terminus compared to the POMGNT1 variants near the 30 terminus (29). For the first time, this study introduces the novel homozygous variant [NM_001243766.2:c.74G>A:p.W25X] within the POMGNT1 gene in hydrocephalus patients, which is indexed with an Accession number “RCV002466682.2” on the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/RCV002466682.2/).
While this study identified a novel pathogenic variant in POMGNT1 associated with isolated congenital hydrocephalus, expanding knowledge in this area, certain limitations must be acknowledged. Assessing the variant’s effect through in vitro functional analyses was beyond the initial scope but remains an important avenue for future investigation. Additionally, segregation of the variant within a single family precludes determining its complete prevalence in unrelated individuals. Furthermore, the study did not investigate other potential genetic causes of hydrocephalus in the family, which could have contributed to the observed phenotype. Addressing these limitations through additional analyses could provide deeper mechanistic insights. Replicating findings in more extensive, diverse cohorts would help establish generalizability. Nonetheless, this work broadened the mutational spectrum and advanced the understanding of hydrocephalus etiology by examining candidate genes through unbiased genomic approaches like WES.
Conclusion
In the present research, the homozygous POMGNT1 variant identified in the proband was novel, resulting in a truncated protein segregating with hydrocephalus in family members. According to the clinical and bioinformatics analysis, this variant was regarded as an NMD with high pathogenicity. In conclusion, the results of this study highlight the destructive potential of variation affecting the buried residue (p.W25) on the neural network and expand the mutational gene spectrum of CH, which can improve understanding of CH etiology and genetics counseling. However, further in vitro functional studies are needed to conclusively determine the molecular consequences and correlate with the observed patient phenotype.
Data availability
Human variant and pertinent phenotype have been reported to ClinVar (Accession Number: RCV002466682.2, https://www.ncbi.nlm.nih.gov/clinvar/RCV002466682.2/).
Abbreviations
- ACMG:
-
American College of Medical Genetics and Genomics
- AC:
-
Abdominal circumference
- AMP:
-
Association for Molecular Pathology
- BAM:
-
Binary Alignment Map
- BPD:
-
Biparietal diameter
- BQSSR:
-
Base quality score recalibration
- CADD:
-
Combined Annotation Dependent Depletion
- CDS:
-
Coding sequence
- CH:
-
Congenital hydrocephalus
- CNV:
-
Copy number variant
- CSF:
-
Cerebrospinal fluid
- DANN:
-
Deep neural network dataset for annotation of deleterious non-synonymous variants
- DAPC:
-
Dystrophin-associated protein complex
- Ensembl VEP:
-
Ensembl variant effect predictor
- ESP:
-
Exome Sequencing Project
- ExAC:
-
Exome Aggregation Consortium
- FATHMM-MKL:
-
Functional analysis through hidden Markov models-Multiple kernel learning
- GATK HC:
-
Genome analysis toolkit Haplotype caller
- GERP:
-
Genomic evolutionary rate profiling
- gnomAD:
-
Genome Aggregation Database
- HC:
-
Head circumference
- HGMD:
-
Human Gene Mutation Database
- HPO:
-
Human phenotype ontology
- IGV:
-
Integrative Genomics Viewer
- Indels:
-
Insertions/deletions
- in silico:
-
By computer or via computer simulation
- IQR:
-
Interquartile range
- in vitro:
-
In artificial environment outside living organism
- MAF:
-
Minor allele frequency
- MDDG:
-
Muscular dystrophy dystroglycanopathy
- MEB:
-
Muscle–eye–brain disease
- mRNA:
-
Messenger RNA
- NGS:
-
Next-generation sequencing
- NMD:
-
Nonsense-mediated mRNA decay
- NTC:
-
No-template control
- PanTHER:
-
Protein analysis through evolutionary relationships
- PCR:
-
Polymerase chain reaction
- PHRED:
-
Program for assigning quality scores
- PMut:
-
Pathogenic mutation prediction
- PVS1:
-
American College of Medical Genetics criteria for variant classification
- PROVEAN:
-
Protein variation effect analyzer
- QC:
-
Quality control
- SAM:
-
Sequence Alignment/Map
- SIFT:
-
Sorting intolerant from tolerant
- SiPhy:
-
Signature of positive selection
- SNVs:
-
Single-nucleotide variants
- UCSC:
-
University of California Santa Cruz
- VEP:
-
Variant effect predictor
- VCF:
-
Variant call format
- WES:
-
Whole-exome sequencing
- WGS:
-
Whole genome sequencing
- WWS:
-
Walker–Warburg syndrome
References
Dewan MC, Rattani A, Mekary R, Glancz LJ, Yunusa I, Baticulon RE et al (2018) Global hydrocephalus epidemiology and incidence: systematic review and meta-analysis. J Neurosurg. https://doi.org/10.3171/2017.10.Jns17439
Estey CM (2016) Congenital hydrocephalus. Vet Clin North Am Small Anim Pract 46(2):217–229. https://doi.org/10.1016/j.cvsm.2015.10.003
Aghayev K, Iqbal SM, Asghar W, Shahmurzada B, Vrionis FD (2021) Advances in CSF shunt devices and their assessment for the treatment of hydrocephalus. Expert Rev Med Devices 18(9):865–873. https://doi.org/10.1080/17434440.2021.1962289
Garcia-Bonilla M, McAllister JP, Limbrick DD (2021) Genetics and molecular pathogenesis of human hydrocephalus. Neurol India 69(Supplement):S268–S274. https://doi.org/10.4103/0028-3886.332249
Duy PQ, Weise SC, Marini C, Li XJ, Liang D, Dahl PJ et al (2022) Impaired neurogenesis alters brain biomechanics in a neuroprogenitor-based genetic subtype of congenital hydrocephalus. Nat Neurosci 25(4):458–473. https://doi.org/10.1038/s41593-022-01043-3
Yamasaki M, Kanemura Y (2015) Molecular biology of pediatric hydrocephalus and hydrocephalus-related diseases. Neurol Med Chir (Tokyo) 55(8):640–646. https://doi.org/10.2176/nmc.ra.2015-0075
Bachmann-Gagescu R, Ishak GE, Dempsey JC, Adkins J, O’Day D, Phelps IG et al (2012) Genotype-phenotype correlation in CC2D2A-related Joubert syndrome reveals an association with ventriculomegaly and seizures. J Med Genet 49(2):126–137. https://doi.org/10.1136/jmedgenet-2011-100552
McCauley J, Masand N, McGowan R, Rajagopalan S, Hunter A, Michaud JL et al (2011) X-linked VACTERL with hydrocephalus syndrome: further delineation of the phenotype caused by FANCB mutations. Am J Med Genet A 155a(10):2370–2380. https://doi.org/10.1002/ajmg.a.33913
Fox NS, Monteagudo A, Kuller JA, Craigo S, Norton ME (2018) Mild fetal ventriculomegaly: diagnosis, evaluation, and management. Am J Obstet Gynecol 219(1):B2-b9. https://doi.org/10.1016/j.ajog.2018.04.039
Morganti S, Tarantino P, Ferraro E, D’Amico P, Duso BA, Curigliano G (2019) Next Generation Sequencing (NGS): a revolutionary technology in pharmacogenomics and personalized medicine in cancer. Adv Exp Med Biol 1168:9–30. https://doi.org/10.1007/978-3-030-24100-1_2
Sullivan W, Reeves BC, Duy PQ, Nelson-Williams C, Dong W, Jin SC et al (2021) Exome sequencing as a potential diagnostic adjunct in sporadic congenital hydrocephalus. JAMA Pediatr 175(3):310–313. https://doi.org/10.1001/jamapediatrics.2020.4878
Maryam N, Kazem P, Sara S, Massoud H (2011) Investigation of exon 1 in FRDA gene in patients with clinically symptomatic Friedreich ataxia. Clin Biochem 13(44):S281
Mousavi NN, Houshmand SM, Naseroleslami M (2020) Novel mutation in Frda gene among Iranian patients with Friedreich's Ataxia
Preiksaitiene E, Voisin N, Gueneau L, Benušienė E, Krasovskaja N, Blažytė EM et al (2020) Pathogenic homozygous variant in POMK gene is the cause of prenatally detected severe ventriculomegaly in two Lithuanian families. Am J Med Genet A 182(3):536–542. https://doi.org/10.1002/ajmg.a.61453
Peiris TJ, Indaram M, Koo E, Soul JS, Hunter DG (2018) Congenital muscular dystrophy-dystroglycanopathy, type A, featuring bilateral retinal dysplasia and vertical angle kappa. J Aapos 22(3):242–4.e1. https://doi.org/10.1016/j.jaapos.2017.12.011
Li M, Fu H, Li J, Meng D, Zhang Q, Fei D (2022) Compound variants of FKTN, POMGNT1, and LAMB1 gene identified by prenatal whole-exome sequencing in three fetuses with congenital hydrocephalus. J Obstet Gynaecol Res 48(10):2624–2629. https://doi.org/10.1111/jog.15358
Binz RL, Tian E, Sadhukhan R, Zhou D, Hauer-Jensen M, Pathak R (2019) Identification of novel breakpoints for locus- and region-specific translocations in 293 cells by molecular cytogenetics before and after irradiation. Sci Rep 9(1):10554. https://doi.org/10.1038/s41598-019-47002-0
Binz RL, Sadhukhan R, Miousse IR, Garg S, Koturbash I, Zhou D et al (2021) Dietary methionine deficiency enhances genetic instability in murine immune cells. Int J Mol Sci. https://doi.org/10.3390/ijms22052378
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–424. https://doi.org/10.1038/gim.2015.30
Spennato P, Mirone G, Nastro A, Buonocore MC, Ruggiero C, Trischitta V et al (2011) Hydrocephalus in Dandy-Walker malformation. Childs Nerv Syst 27(10):1665–1681. https://doi.org/10.1007/s00381-011-1544-4
Krejčí O, Krejčí T, Mrůzek M, Večeřa Z, Šalounová D, Lipina R (2021) Hydrocephalus caused by primary fourth ventricle outlet obstruction: our experience and literature review. World Neurosurg 148:e425–e435. https://doi.org/10.1016/j.wneu.2021.01.008
Mastromoro G, De Luca A, Marchionni E, Spagnuolo A, Ventriglia F, Manganaro L et al (2021) External hydrocephalus as a prenatal feature of noonan syndrome. Ann Hum Genet 85(6):249–252. https://doi.org/10.1111/ahg.12436
Rufus P, Moorthy RK, Joseph M, Rajshekhar V (2021) Post traumatic hydrocephalus: incidence, pathophysiology and outcomes. Neurol India 69(Supplement):S420–S428. https://doi.org/10.4103/0028-3886.332264
Akasaka-Manya K, Manya H, Kobayashi K, Toda T, Endo T (2004) Structure-function analysis of human protein O-linked mannose beta1,2-N-acetylglucosaminyltransferase 1, POMGnT1. Biochem Biophys Res Commun 320(1):39–44. https://doi.org/10.1016/j.bbrc.2004.05.129
Hohenester E (2019) Laminin G-like domains: dystroglycan-specific lectins. Curr Opin Struct Biol 56:56–63. https://doi.org/10.1016/j.sbi.2018.11.007
Diesen C, Saarinen A, Pihko H, Rosenlew C, Cormand B, Dobyns WB et al (2004) POMGnT1 mutation and phenotypic spectrum in muscle-eye-brain disease. J Med Genet 41(10):e115. https://doi.org/10.1136/jmg.2004.020701
Kuwabara N, Manya H, Yamada T, Tateno H, Kanagawa M, Kobayashi K et al (2016) Carbohydrate-binding domain of the POMGnT1 stem region modulates O-mannosylation sites of α-dystroglycan. Proc Natl Acad Sci USA 113(33):9280–9285. https://doi.org/10.1073/pnas.1525545113
Manya H, Kuwabara N, Kato R, Endo T (2020) FAM3B/PANDER-like carbohydrate-binding domain in a glycosyltransferase, POMGNT1. Methods Mol Biol 2132:609–619. https://doi.org/10.1007/978-1-0716-0430-4_52
Taniguchi K, Kobayashi K, Saito K, Yamanouchi H, Ohnuma A, Hayashi YK et al (2003) Worldwide distribution and broader clinical spectrum of muscle-eye-brain disease. Hum Mol Genet 12(5):527–534. https://doi.org/10.1093/hmg/ddg043
Acknowledgements
We wish to thank all our colleagues in Molecular Research Center and Department of Cellular and Molecular Biology, Faculty of Advanced Science and Technology, Tehran Medical Sciences, Islamic Azad University, Tehran, Iran. Authors appreciate their coworkers and patients who kindly contributed to this research. The authors are especially thankful to the personnel of the Noorgene Genetic & Clinical Laboratory (http://www.noorgene.com/).
Funding
This research was financially supported by a grant from vice-chancellor for research affairs of Tehran Medical Sciences, Islamic Azad University, Tehran, Iran. This paper is issued from the thesis of Masoud sabzeghabaiean, MSc student of Cell and Molecular Biology.
Author information
Authors and Affiliations
Contributions
Dr. MN conceived the manuscript and revised it. MS and MM wrote the manuscript, and HK prepared tables. Dr. FG and ZZ were included in qualifying the concept and design. Dr. FG also investigated cellular and biological mechanisms. Dr. JM and MM critically evaluated the intellectual contents and analyzed the results of genetic tests. All authors participated in preparing the final draft of the manuscript, revised the manuscript, and critically assessed the academic contents. Also, all authors have read and approved the manuscript’s content and confirmed the accuracy or integrity of any part of the work.
Corresponding author
Ethics declarations
Human and animal participants
All the procedures performed in the studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
The Islamic Azad University, Tehran Medical Sciences Branch, approved the study, and all the experiments were conducted following the standard ethical protocols of the institutional and national research committee. Informed consent was obtained from all participants in this investigation and parents as the proband’s legal guardians to publish any potentially identifiable data in this article.
Competing interests
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sabzeghabaiean, M., Maleknia, M., Mohammadi-Asl, J. et al. The homozygous pathogenic variant of the POMGNT1 gene identified using whole-exome sequencing in Iranian family with congenital hydrocephalus. Egypt J Med Hum Genet 25, 38 (2024). https://doi.org/10.1186/s43042-024-00513-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-024-00513-6