
Abstract
Introduction
Ullrich congenital muscular dystrophy (UCMD) is a severe form of inherited muscle weakness at birth. Recent genetic studies discovered that different gene mutations are responsible for UCMD clinical manifestation.
Case report
In this study, we carried out whole exome sequencing (WES) to recognize probable gene defects in an Iranian boy with UCMD. We found a novel disease-causing COL6α1 gene mutation (c.2551_2562del; p.Phe851_Arg854del), located in exon35 (NM_001848.3), causing a deletion mutation that has eliminated 12 bp. The WES-identified variant that was confirmed by Sanger sequencing for the patient and his consanguineous parents. Here, we report the clinical manifestations of 4-year-old Iranian patient who presented with muscle weakness since birth and proved compound homozygous mutation of the COL6A1 gene.
Conclusion
Our findings established that this detected COL6α1 mutation is the pathogenic variant for UCMD. This is the first genetic study indicating that c.2551_2562 mutation in homozygous state in COL6α1 gene is responsible for the UCMD phenotype.
Similar content being viewed by others


Introduction
Ullrich Congenital Muscular Dystrophy (UCMD) is a rare genetic disorder defined by inherited collagen type VI deficiency related to myopathy. It was first described in 1930 by Otto Ullrich. The disease manifests itself with a combination of hypotonia at birth, proximal joint contractures and distal joint hyperlaxity (hypermobility), and severe generalized muscle weakness.
In 80% to 99% of patients. UCMD is a severe form with an autosomal recessive disorder, with an estimated prevalence of 0.13 per 100,000 in England. There is a milder form known as Bethlem Myopathy (BM) caused by autosomal dominant mutations. BM was initially reported in 1976 by Bethlem and Wijngaarden, with an estimated prevalence of 0.77 per 100,000 [1].
Studies have shown that muscular dystrophies are caused by a mutation in genes. These mutations may be passed on from parents to children or they may occur denovo in an individual.
Based on previous studies, the disorder may vary in outcome and severity of pathological finding. Classical BM (MIM#158810) conforms to a milder form with late-onset and static course. The differential spectrum of these two contiguous diseases is determined in muscle biopsy. Other tissues in patients with UCMD are also involved, reflecting the wide distribution of Collagen type VI and its variety.
The clinical symptoms observed in UCMD are mobility impairments caused by muscle weakness and hyperlaxity. The affected patient can take advantage of physiotherapy to promote movement abilities [2].
Nevertheless, using a standing frame may be helpful to attain vertical posture or to postpone the development of scoliosis and contractures in proximal joints and distal joints hyperlaxity. Respiratory failure is the main cause of death at 1 or 2 decades.
Previously, it has been identified as a genetic abnormality in the group of genes including ColVI (α1_α2_α3) associated with UCMD and BM cases. Collagen TypeVI is a microfibrillar collagen which is produced by mesenchymal stromal cells formed from dimers stabilized by disulfide bonds. Finally, tetramers are generated by parallel alignment in the outer rod-like region which overlap at the globular end domains, connected and secreted to the extracellular matrix (ECM) where they are incorporated to organize a network (beaded microfilaments with a typical 100 nm frequency) that includes N, C terminals and shows similarity with VWF-A domains.
As ubiquitous proteins in extracellular matrix, Collagen typeVI is a widespread heterotrimeric glycoprotein throughout the ECM that complex of COL6 (α1_α2_ α3_α4_α5_α6). The α1 and α2 chains of Collagen typeVI are similar and encoded by single-copy gene in close proximity on human chromosome 21q22.3 a gene-rich region that has proved to refractory cloning. COL6α3 is in 2q37 [2, 3], COL6α4_α5 are homologue to α3 but they represent a difference in pattern. Col6α4 due to large chromosomal inversion separated into two pieces (COL6α4P1 and COL6α4P2) Col6α4P1 code a protein while COL6α4P2 has been regarded as Pseudogene which does not code a protein but may be involved in regulating the expression of Col6α4P1. In muscle, collagen and the protein called Perlecan bind muscle cells to the ECM [2].
COL6α1 encoded by total of 36 exon coating approximately 30 KB. The helical chains collected by repeating (Gly_x_y) motif within the TH domain of COL6α1 and entails Cys residues, the most mutations are occurred in BM caused interrupt Gly_x_y repeating sequence by glycine substitution [2, 3].
Moreover, the complex of Collagen type VI is significant to maintain the elasticity of blood vessels and supply flexibility and resilience of joints, to promote adhesion and to interact with integrins, and it may also regulate growth factors. Imperfect structure and low-level production of Collagen type VI could disrupt the connections between ECM, connective tissues, skin, tendon, cartilage, skeletal muscle, peripheral nervous system, and intervertebral discs.
UCMD (MIM#254090) results from a homozygous or heterozygous pathogenic variant which is skipping one of the 3 genes encoded the subunits of Collagen type VI [4, 5].
Case report
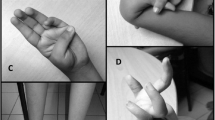
We have enrolled an Iranian patient, who suffered from UCMD in Ahvaz (Khuzestan, province, Iran). The patient, a 4-year-old male, was born following an uneventful pregnancy. He had delayed closure of the fontanels, reduced fetal movements, hypotonia at birth. Muscle weakness was observed around the age of 2-years and progressively worsened with increasing age. He needed aid to stand, lift the trunk and he had difficulty to raise devices, he demonstrated difficulty in standing up but he was able to walk independently and did not show any recorded sign of respiratory failure. His cognitive language and social development were age appropriate. Clinical examination showed bilateral proximal muscle weakness in both upper and lower extremities, hypotonia and hyperlaxity of the distal joints was noted involving the wrists and fingers. During the examination, Deep Tendon Reflexes (DTR) were graded as 2 (normal reaction), Nerve Conduction test (NCS) was performed by an neurologist. The patient had no PNS involvement and sensorial, motor, and mental status were totally normal which were noted in patient records. Hinge on the biochemical test Serum creatine kinase (CK) levels were elevated, muscle magnetic resonance imaging (MRI) was done and revealed lower extremities, high intensity on T1-weighted imaging was observed in the peripheral area, which revealed clear involvements in the trunk. Rectus femoris contracture was obvious. Contracture in pelvic girdle and spine along with proximal joint contractures and hyperlaxity of distal joints were also reported.
In this study, which was approved by the Noor Gene Genetic Lab, Ahvaz, Iran and written informed consent was obtained from the parents granting permission for genetic analyses, a genomic sequence of COL6A1 was performed. We obtained peripheral blood samples with informed consent and analyzed DNA samples of the index patient. The gDNA was extracted from peripheral leukocytes of each participant using the salting standard method and polymerase chain reaction (PCR) amplification of the 1 coding exon (exon35) of Col6α1 was conducted by using oligonucleotide primer pairs.
Whole Exome Sequencing (Macrogen, Seoul, South Korea) was conducted to detect mutation in particular genes. This type of hereditary dystrophy disease in gDNA of the proband has been identified by molecular genetic analysis, which is displayed as an autosomal inheritance model with a homozygous del mutation with eliminated 12 bp.
Total RNA was isolated from skin fibroblasts of the patient, using amplification primers within exons 35 of the COL6A1 gene Real-Time PCR was performed on cDNA from fibroblasts, using TaqMan expression assays for the three COL6A genes. Real-Time PCR showed a vigorous reduction in the aberrant COL6A1 transcripts with non-mutated COL6A2 and COL6A3 mRna. There was a marked variation in fiber size. Examination by transmission electron microscopy revealed short collagen VI microfibrils, which became clear unable to expand regular webs (Fig. 1).

Consanguineous pedigree with COL6α1 mutation demonstrating our effected patient, the patients are denoted in black. Healthy mother and sister, carrier father, and the filled symbols in elder siblings indicate affected individuals
Short collagen VI fibrils, organized by parallel microfibrils, were sporadically found, and the globular domains of adjoining microfibrils were not regularly aligned. Fibers with internalized nuclei were scattered although no necrotic or regenerating fibers were observed. Mild endomysial fibrosis were observed ([H&E] staining).
Consequently, in this study, we discovered a novel del mutation in the COL6α1 gene similar to previous del mutations associated with UCMD (Table 1).
A novel homozygous deletion mutation, likely pathogenic variant in exon35 of the COL6A1 gene NM_001848.3:c.2551_2562 (p.851_854del) identified in proband.
Sanger sequencing verified deletion in (p.Phe851_Arg854del), no mutation was detected on other genes.
Based on the American College of Medical Genetics and Genomics (ACMG) guidelines, the c.2551_2562del mutation was classified as likely pathogenic with an autosomal recessive phenotype. The patient was born of consanguineous parentage who carried a heterozygous form of the mentioned deletion (Fig. 2)

Electropherogram of the affected patient and his carrier parents demonstrating a homozygous c.2551_2562del mutation in the COL6α1 gene and heterozygous carrier state in his parents
Discussion
UCMD and BM are characterized by muscle weakness that represents the opposite end of ColVI related myopathy from BM with static course to UCMD with severe course observed by Electromyography (EMG) [7].
Till this point in time, no treatment has been introduced to COL6A related muscular dystrophy, the symptoms are just managed as we cited before. Recent research in the mice model of collagen typeVI deficiency (Col6α1−/−) shows that CyclosporineA can revive at least part of affected tissue caused by mitochondrial deficiency [8].
Collagen typeVI deficiency is caused by a stop codon at the genome or frameshift including insertion, deletion, duplication, and splice changes, which could lead to mitochondrial defect (mitochondrial abnormality spanning from tubular cristae to electron-dense matrix with alteration of threshold voltage for PTP opening caused by F1F0-ATPase Inhibitor oligomycin), sarcoplasmic reticulum, decreased autophagy (a crucial reaction against muscle wasting), impaired muscle regeneration, and variation in fiber size atrophy, which are mostly replaced by fibrotic tissue in the diaphragm, and finally ending in maturation deficiency [8, 9].
Besides, in 80 to 99% of symptoms that follow these alterations are due to muscle weakness in all four limbs that could result in limited or loose mobility before adulthood.
And by according to in terms of MRI findings, collagen typeVI related to myopathy is characterized by fatty Infiltration, contracture of spine, hyperlaxity of distal joints (finger, wrist, toes), and hyperlaxity of axial and proximal joints (hips, shoulder, knees). This sign was observed in two brothers in upper and lower limb extremities caused by pathogenic variant in exon25, NM_001848:c.1667G > T;NP_001839.2:p.Gly556Val [10, 11].
Reduction in paxillin and PI3K signaling, owing to reduced activation of CDC42 when mutations occur in collagen typeVI leads to respiratory failure, bronchopneunemonia, and diaphragmatic paresis and is a common cause of death at the first or second decade of life [12].
Changes in skin by follicular keloids or atrophic scar formation (keratosis pilaris associated with atopy its common skin condition in ColVI deficiency) [1], are associated with hypotonia at birth and tight chilies tendon. Mutation (c.227 + 2 T > C) in the COL6A1 gene causes torticholis, cervical, and lumbar spine rigidity, which may be due to asymmetric contracture in mm. scalenii, and m. sternocleidomastoideus [13].
In contrast to previous studies, the most complications in our proband were in trunk and pelvic girdle and didn’t show any involvement in PNS leading to spinal cord, or any symptoms in skin that was mentioned in the previous study.
Hence, in this study, we reported a 4-year-old boy with a pathogenic mutation in COL6α1 related myopathy. We initially focused on the analysis of the UCMD proband who exposed a compound homozygosis for autosomal recessive COL6α1 mutation. A deletion within exon35 NM_001848.3:c.2551_2562del:p.Phe851_Arg854 eliminated 12 bp. Genetic testing was performed through whole-exome sequencing (WES).
WES and PCR analysis for the UCMD patient revealed pathogenicity of novel mutation categorized in developmental delay motor. The pattern of inheritance is autosomal recessive because the patient carried the homozygous mutation and his parents were heterozygous for the detected mutation.
Conclusion
At the end of this case study, we detected a case of UCMD with a novel homozygous COL6α1 gene mutation (c.2551_2562del p.Phe851_Arg854) in an Iranian boy from a heterozygous and carrier parents. It can be concluded that the discovered mutation is a novel one that was carried in patient’s ancestors using WES technique. In addition, similar to previous reports, our study evidenced that mutation in the COL6α1 gene is associated with UCMD and BM.
Availability of data and materials
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Saroja AO et al (2013) Bethlem myopathy: an autosomal dominant myopathy with flexion contractures, keloids, and follicular hyperkeratosis. Ann Indian Acad Neurol 16(4):712
Weil D, Mattei MG, Passage E, N’Guyen VC, Pribula-Conway D, Mann K et al (1988) Cloning and chromosomal localization of human genes encoding the three chains of type VI collagen. Am J Hum Genet 42:435–445
Giusti B et al (2005) Dominant and recessive COL6A1 mutations in Ullrich scleroatonic muscular dystrophy. Ann Neurol 58(3):400–410
Bernardi P, Bonaldo P (2013) Mitochondrial dysfunction and defective autophagy in the pathogenesis of collagen VI muscular dystrophies. Cold Spring Harb Perspect Biol 5(5):a011387
Mercuri E et al (2002) Collagen VI involvement in Ullrich syndrome: a clinical, genetic, and immunohistochemical study. Neurology 58(9):1354–1359
Zhang YZ et al (2014) Novel collagen VI mutations identified in Chinese patients with Ullrich congenital muscular dystrophy. World J Pediatr 10(2):126–132
Peat RA et al (2007) Variable penetrance of COL6A1 null mutations: implications for prenatal diagnosis and genetic counselling in Ullrich congenital muscular dystrophy families. Neuromuscul Disord 17(7):547–557
Mereness JA, Mariani TJ (2021) The critical role of collagen VI in lung development and chronic lung disease. Matrix Biol Plus 10:100058
Williams L, Layton T, Yang N, Feldmann M, Nanchahal J (2022) Collagen VI as a driver and disease biomarker in human fibrosis. FEBS J 289(13):3603–3629
Lamandé SR et al (1999) Bethlem myopathy and engineered collagen VI triple helical deletions prevent intracellular multimer assembly and protein secretion. J Biol Chem 274(31):21817–21822
Williams L, Layton T, Sirisena ND et al (2021) A novel variant in the COL6A1 gene causing Ullrich congenital muscular dystrophy in a consanguineous family: a case report. BMC Neurol 21(1):1–7
Allamand V et al (2010) 166th ENMC international workshop on collagen type VI-related myopathies, 22–24 May 2009, Naarden, The Netherlands. Neuromuscul Disord 20(5):346–354
Bardakov SN et al (2021) Intrafamilial phenotypic variability of collagen VI-related myopathy due to a new mutation in the COL6A1 gene. J Neuromuscul Dis 8(2):273–285
Acknowledgements
We wish to thank the family members for their participation in this study.
Funding
None.
Author information
Authors and Affiliations
Contributions
EN made design of the study and wrote the manuscript. EN, ESHY, JMA analyzed and interpreted the data. JMA edited the manuscript. ZMM helped to review the manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Written informed consent was obtained from the parents of the patient. All procedures performed in this study were in accordance with the ethical standards of the institutional and/or national research committee.
Consent for publication
Written informed consent was obtained from the family for this publication.
Competing interests
The authors declared there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nekouei, E., Yancheshmeh, E.S., Mohammadi-Asl, J. et al. Exome sequencing identified a novel Col6α1 mutation in an Iranian patient with Ullrich congenital muscular dystrophy: a case report. Egypt J Med Hum Genet 23, 166 (2022). https://doi.org/10.1186/s43042-022-00372-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-022-00372-z