
Abstract
Background
The B-cell prolymphocytic leukemia (B-PLL) diagnosis is challenging due to the superposition with mature B-cell leukemia and/or lymphoma.
Objective
An insight case study of trisomy 5 with complex karyotype abnormalities in B-cell prolymphocytic leukemia.
Subject and methods
A 72-year-old man was referred to the Hematology Department, Qilu Hospital, Shandong University, because of persistent fever (10 days) and lymphocytosis. A detailed diagnostic methods including complete blood count, bone marrow aspiration, flow cytometry, conventional karyotype analysis, fluorescence in situ hybridization (FISH), quantitative real-time polymerase chain reaction (qRT-PCR), next-generation sequencing technology (NGS) used to detect 41 kinds of mutant genes related to hematological malignancies were conducted and reasonable therapeutic regimens including emergent leukapheresis accompanied by basification of urine and hydrotherapy, followed by a regimen of cyclophosphamide and dexamethasone.
Results
Subject white blood cell count was 143.43 × 109/L, and 56% prolymphocytes. He did not show lymphadenopathy but splenomegaly. Immunophenotyping of prolymphocytes was CD5(+low), CD10(−), CD11c(−), CD19(+), CD20(+), cCD22(+), CD23(−), cCD79a(+), CD79b(+), FMC7(±), CD43(−), CD3(−), CD56(−), CD103(−), HLA-DR(+), and Lambda(+). R-banding and FISH revealed that leukemia cells carried extra chromosome 5. Considering the rare occurrence of trisomy 5 found in prolymphocytic leukemia, especially in Asians, with rapid disease progression. We know that median survival of B-PLL is three years after diagnosis, while survival time of this patient was only 1 month.
Conclusion
This study could provide the firsthand materials for precision, medicine and mechanism research in cytogenetics and molecular biology. It inferred that trisomy 5 might be a poor prognosis indicator, providing directions for clinical practice in the foreseeable future.
Similar content being viewed by others
Introduction
B-cell prolymphocytic leukemia (B-PLL) is an aggressive, out of control growth of B- lymphocyte cells with scarce neoplasm that represents 1% of lymphocytic leukemia and accompanied by a low survival rate. B-PLL was first described as a variant form of B-cell chronic lymphocytic leukemia (CLL) in the early 1970s [1,2,3,4,5,6,7], while it was implicated as an eminent clinic-pathologic entity and realized as a prolymphocytes malignancy influencing bone marrow (BM), blood, and spleen in the fourth edition of the 2008 World Health Organization (WHO) categorization [2,3,4,5,6,7,8,9]. 69 years is the mean age at diagnosis, with slightly male predominance. Patients typically exhibit B-symptoms, lymphocytosis, and splenomegaly, but lymphadenopathy is infrequent. It is considered by the WHO when more than 55% of the peripheral blood the lymphocytes are prolymphocytes, or when a lymph node or BM sample shows that prolymphocytes are the majority of lymphocytes. Molecular profiling and cytogenetic studies have failed to define and represent a single genetic aberration, instead abnormality of TP53/p53, deletion on 11q23, and/or 13q14, and trisomy 12 complex karyotypes have been documented. It is particularly worth mentioning that patient with karyotype abnormalities involving trisomy 5 is the first time to be reported in prolymphocytic leukemia [3,4,5,6,7,8,9,10,11].
Case report
A 72-year Chinese male was admitted to our Hematology Department, Qilu Hospital, Shandong University, with a history of 10-day fever, fatigue, poor appetite, diarrhea, and abdominal pain. His previous medical history showed that he had a chronic hepatitis B for almost 40 years and painless splenomegaly for about ten years. He was a peasant and had no reported exposure to environmental carcinogen. On the admission day, he presents a brief syncopal episode. Upon inspection, he had a 38.3 °C temperature, conjunctiva and mucosal pallor was reported. The superficial lymph nodes were not palpable. About 7 cm below the left costal margin the spleen tip was palpable and confirmed through radiological investigations including: chest x ray, and abdominal ultrasound as conducted in previous studies and published leukemia diagnosis work [6, 10, 12]. Rest of the physical examinations was negative. The whole blood count revealed the white cell, hemoglobin, and platelet were 143.43 × 109/L, 101 g/L, and 79 × 109/L, respectively. The peripheral blood smear showed 87% of the nucleated cells were leukemic blasts with an elevated nuclear/cytoplasmic ratio, 56% of which were prolymphocytes with larger cell bodies and more prominent nucleoli. Renal and hepatic functions were normal. qRT-PCR displayed that serum level of Hepatitis B DNA and lactate dehydrogenase (LDH) was 1.25 × 103 IU/mL and 2014 U/L respectively, which was much higher than normal which could be also be related to post chemotherapy death. The current work was approved by Shandong University Research Ethical Committee, and written informed consent was obtained from participant family enrolled prior to sample collection.
Morphology and immunology
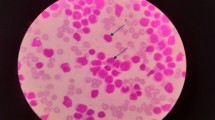
Bone marrow aspiration, smear and biopsy were carried out for morphological and immunological studies according to previous studies [6, 10,11,12,13]. The bone marrow smear represented 100% cellularity, with 91% blasts infiltration. Sixty-five percent among them were prolymphocytes characterized by large cell bodies, and prominent nucleoli. The blasts exhibit a high ratio of nuclear versus cytoplasm. Auer rods were not present (Fig. 1). Flow cytometry showed the blasts expressed CD19, CD20, CD5 (low), CD79a, cCD22, cCD79a, HLA-DR, FMC7 (low), and Lambda, but did not express CD10, CD23, CD34, myeloperoxidase (MPO), cCD3 and Kappa. All results above revealed the patient with mature B-cell neoplasm, in which B-PLL, mantle cell lymphoma (MCL), a variant form of hairy cell leukemia (HCL-V) and splenic marginal zone lymphoma (SMZL) were highly suspicious types (Fig. 1).

The result of bone marrow morphology of B-cell prolymphocytic leukemia [() prolymphocytes are medium-sized lymphoid cells with a round nucleus, moderately condensed nuclear chromatin, a prominent central nucleolus, and basophilic cytoplasm]
Cytogenetics
Cells were cultured in penicillin (100U/mL), streptomycin (100 fig/mL), and 10% fetal calf serum (FCS) in the presence of TPA (0.05 pg/mL final concentration) RPMI. Cells were harvested after 3 and 5 days of culture, and treated with hypotonic solution (075 mol/L, KCI0, 10 min, 37 °C) after (0.1 pg/mL colcemid, 60 min) mitotic arrest and with acetic acid/methanol (l:3) fixed. Metaphase chromosomes G banded were applied according to universal methods.
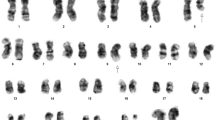
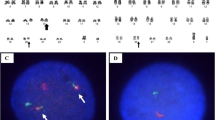
Fluorescence in situ hybridization (FISH) analysis: hypotonically fixed cells were placed drop-wise on slides and with fluorescent probes against 5q, 7q, 20q12, + 8, − Y, RB1, 1q21, D13S319 and IgH hybridized for 16 h. Under a fluorescence microscope cells were observed after staining with DAPI, the for fluorescence signals measurement. FISH was used to detect cytogenetic abnormalities. According to FISH results, three red, and three green fluorescent signals were observed by using two pairs of probes, CSF1R/D5S23, D5S7213(5q33) and EGR1/D5S23, D5S721(5q31), which were located in 5, that means it has an extra trisomy 5 (Fig. 2) and the detection of t (11;14) (q13; q32) translocation by CCND1/IGH probe was negative. We also used other probes, which are located in 7q31, 20q12,+8, ATM (11q22.3), RB1 (13q14), D13S25 (13q14.3), P53 (17p13.1), IGH, D13S319, and 1q21 that associated with chromosomal changes of CLL, MM, and MDS, but there was no positive result. The Karyotype analysis was performed using a standard protocol. It revealed that 49, XY,+ 5, del(5)(q13), add(8)(p23),+ 15, add(17)(q25),+19[cp20] (Fig. 3), which was the same as the FISH results (Figs. 2, 3).

The FISH analysis results with red (color) and green (color) probing of interphase B-cell prolymphocytic leukemia displaying a normal fusion signal and b split signal (arrow) red 5' centromeric region and (arrow) green 3' telomeric region

The R-banded complex karyotype results representing the new B-cell prolymphocytic leukemia trisomy of 15, 19 and 5 (arrow)
Molecular biology
qRT-PCR was utilized to determine the molecular abnormality in bone marrow cells from the patient. However, we did not observe any positive result on 43 kinds of fusion genes detection, such as breakpoint cluster region/Abelsen (BCR/ABL), promyelocytic leukemia/retinotic acid receptor alpha (PML/RARA), a core-binding factor β myosin heavy chain11 (CBFB/MYH11), acute myeloid leukemia 1/eight-twenty one (AML1/ETO), mixed-lineage leukemia (MLL) gene rearrangement (Table 1). Genomic material was extracted for targeted gene mutation panel by NGS technology. Bioinformatics and statistics were used to determine the association between clinical features and gene mutations also discuss of our results with the Cancer Genome Atlas Research Network (TCGA) public dataset. NGST was used to detect 41 kinds of mutant genes related to hematological malignancies, and the results showed mutation rates of SETBP1 and PML were 51.06% and 98.54% (Table 2). PCR determination of B cell clonal immunoglobulin gene rearrangements showed the positive result of B cell monoclonality.
Treatment
The patient of study was diagnosed with B-cell prolymphocytic leukemia. He underwent emergent leukapheresis accompanied by basification of urine and hydrotherapy. Then, we started on a regimen of cyclophosphamide and dexamethasone instead of fludarabine, considering the patient and his family conditions, including their poor economic incomes and the patient’s age. After 10-day therapy, the patient attained slightly stable and improving conditions with normal temperature, significantly decreased white cell count and safe levels of hemoglobin and platelet. He left our hospital against medical advice without continuous supportive treatment, and observation. He died at the local hospital 11 days after leaving our hospital. The family declined an autopsy. No further studies can be performed due to the patient’s death.
Discussion
We described a case of B-PLL carrying trisomy 5 with complex karyotype abnormalities, which was determined by conventional karyotype analysis and interphase/metaphase FISH. It is known that the B-PLL diagnosis is challenging because of superposition with MCL, HCL-V, or SMZL mature B-cell leukemia and lymphoma. The 72-year-old patient exhibited the typical features of B-PLL, lymphocytosis, splenomegaly, and B-symptoms. Though the patient was old, the results of morphology and immunology detection, as well as rapid and marked increase in the peripheral white cell count, were observed, which were both different from the features of CLL. The diagnosis requirements of more than 55% of the lymphocytes in the peripheral blood are prolymphocytes, medium-sized cells with prominent central clumped chromatin nucleoli, and typical immune-phenotype, the expression of the B-cell antigens CD19, 20, 22 and 79a were present [8, 9]. Moreover, the absence of t(11;14)(q13;q32) helped exclude MCL. Smooth outline of the blast cells, B-symptoms, higher WBC (> 100 × 109/L), and clinical course much more attributes of B-PLL compared to HCL-V or SMZL. Research in sixty subjects showed that splenomegaly was the most prevalent feature of B-PLL [7]. However, the molecular pathogenesis of B-PLL is still largely unknown. As for studies reported, complex karyotype and abnormalities in the TP53 tumor suppressor gene might be common events in B-cell PLL [9,10,11,12]. Moreover, deletions at 11q23, 13q14, and trisomy (especially 12) were found [11, 14,15,16,17]. A few case studies have reported transformed B-PLL and CLL, MYC translocations, including t(8;14) [11, 12, 16,17,18].
Because gene fusions are crucial factors for risk stratification in leukemia risk-adapted therapy, gene fusion is indispensable diagnostic testing part. In the present work, fusion gene panels and clinical utility were analyzed based on targeted NGS for leukemia diagnosis. 41 kinds of mutant genes related to hematological malignancies, and the results showed mutation rates of SETBP1 and PML were 51.06% and 98.54% (Table 2). PCR determination of B cell clonal immunoglobulin gene rearrangements showed the positive result of B cell monoclonality [19, 20].
Unfortunately, we did not carry on an optimal individualized treatment regimen considering his economic income and their wishes. However, the choice of personalized treatment regimen is always the critical point in research of hematological disorders. A study on four patients with PLL was carried out, whose age ranged between 55 and 84 years, with the leucocyte count above 180/nL, β2-microglobulin and LDH higher with a median of 4.2 mg/L and284 U/L, respectively. The results showed that 4 patients achieved a complete remission with immune-chemotherapy fludarabine, epirubicin, and rituximab (FER regimen). It provided an impetus for further evaluation of the treatment schedule offered in a larger number of PLL patients [16,17,18,19,20,21]. Previous reports report the successful B-PLL treatment with monotherapy rituximab, while the durability responses were short [16,17,18,19,20,21,22]. All in all, integration of rituximab together with bendamustine or fludarabine with epirubicin or mitoxantrone (FER, BMR, and FMR) as anthracycline has been showed to be efficient in B-PLL [16, 22,23,24,25,26]. Some experts suggest that to patients with no TP53 abnormalities the usual approach is applying this regimen as cornerstone therapy. Alemtuzumab is vital in the treatment for B-PLL patients with mutations and/or deletions of TP53. Also, antagonists targeting molecules for the B-cell receptor as PI3 Kinase delta (PI3Kd) and Burton’s Tyrosine Kinase, may also have efficacy in B-PLL, including TP53 abnormalities cases. However, ofatumumab, GA101, or new anti-CD20 mAbs have not been assessed in B-PLL. Recent researches revealed that idelalisib, a powerful oral and selective PI3Kd inhibitor, combined with rituximab responses was found to be clinically meaningful and rapid in B-PLL patients with disrupted TP53 [21,22,23,24,25,26,27]. Also, Gordon et al. stated that two B-PLL patients, with del(17p), responded to ibrutinib single therapy, an oral inhibitor of Burton’s tyrosine kinase, involved in signal transduction of B-cell receptor. Two patients treated with ibrutinib for months reached normal absolute count of lymphocyte, hemoglobin and platelet [26,27,28,29,30,31,32,33,34,35,36]. At last, the only therapeutic approach in B-PLL and should be taken into account for younger subjects remain the allogeneic hematopoietic stem cell transplant (allo-HSCT).
Conclusion
In conclusion, complex karyotypic abnormalities and aberrations in TP53 probably indicated the B-PLL poor prognosis. Even deficiency of efficient established therapeutic modalities, FMR, FER, BMR, alemtuzumab, idelalisib-rituximab and ibrutinib all exhibited remarkable effect on therapy of B-PLL patients. Taking the cost of treatment into account, more economical and effective regimens should be researched to make sure that patients can afford them. Further molecular profiling and cytogenetic studies remain to be performed to define genetic aberration clearly, such as trisomy 5. While we did not demonstrate the role of trisomy 5 on B-PLL, we put emphasis on specific cytogenetic and molecular abnormalities, which could promote the deep-going research in the field of B-PLL.
Availability of data and materials
Data and materials are available upon request.
Abbreviations
- B-PLL:
-
B-cell prolymphocytic leukemia
- FISH:
-
Fluorescence in situ hybridization
- qRT-PCR:
-
Quantitative real-time polymerase chain reaction
- NGS:
-
Next-generation sequencing
- CLL:
-
Chronic lymphocytic leukemia
- BM:
-
Bone marrow
- LDH:
-
Lactate dehydrogenase
- BCR/ABL:
-
Breakpoint cluster region/Abelsen
- PML/RARA:
-
Promyelocytic leukemia/retinotic acid receptor alpha
- CBFB/MYH11:
-
Core-binding factor β myosin heavy chain11
- AML1/ETO:
-
Acute myeloid leukemia 1/eight-twenty one
- MLL:
-
Mixed-lineage leukemia
- WHO:
-
World Health Organization
References
Swerdlow SH, Campo E, Harris NL (eds) (2017) WHO classification of tumours of haematopoietic and lymphoid tissues, 4th edn. IARC Press, Lyon
Melo JV, Catovsky D, Galton DA (1986) The relationship between chronic lymphatic leukemia and prolymphocytic leukaemia, I: clinical and laboratory features of 300 patients and characterization of an intermediate group. Br J Haematol 63(2):377–387
Dearden C (2012) How I treat prolymphocytic leukemia. Blood 120(3):538–551
Dearden C (2015) Management of prolymphocytic leukemia. Hematol Am Soc Hematol Educ Program 2015:361–367
Collignon A, Wanquet A, Maitre E (2017) Prolymphocytic leukemia: new insights in diagnosis and in treatment. Curr Oncol Rep 19(4):29
El-Kaream SAA, Ebied SAEM, Sadek NA, Attia KAH, Nadwan EA (2021) Serum estrogen and its soluble receptor levels in Egyptian patients with chronic myeloid leukemia: a case-control study. Indian J Hematol Blood Transfus. https://doi.org/10.1007/s12288-021-01451-8
Cross M, Dearden C (2019) B and T cell prolymphocytic leukemia. Best Pract Res Clin Haematol 32(3):217–228
Wandt H, Haferlach T, Thiede C et al (2010) WHO classification of myeloid neoplasms and leukemia. Blood 115(748–9):749–750
Pittman S, Catovsky D (1983) Chromosome abnormalities in B-cell prolymphocytic leukemia: a study of nine cases. Cancer Genet Cytogenet 9:355–365
Ebied SAE, Sadek NA, Zaki NES, Abd El-Kaream SA, El Kashif HKA (2018) Prognostic value of soluble angiotensin II receptor 1 and soluble angiotensin converting enzyme [CD 143] in patients with acute leukemia. Acta Haematol Pol 49(4):240–250. https://doi.org/10.2478/ahp-2018-0028
Bindra BS, Kaur H, Portillo S, Emiloju O, Garcia de Jesus K (2019) B-cell prolymphocytic leukemia: case report and challenges on a diagnostic and therapeutic forefront. Cureus 11:e5629
El-Kaream SAA, Ebied SAEM, Sadek NA, Saad DM, Nadwan EA (2021) Serum estrogen and its soluble receptor levels in Egyptian patients with acute leukemia: case-control study. Egypt J Med Hum Genet 22:68. https://doi.org/10.1186/s43042-021-00186-5
Helaly NA, Esheba NE, Ammo DEA, Elwan NM, Elkholy RA (2021) High Bax/Bcl-2 ratio is associated with good prognosis and better survival in patients with B cell chronic lymphocytic leukemia. Leuk Res 107:106604
Lens D, De Schouwer PJ, Hamoudi RA (1997) p53 abnormalities in B-cell prolymphocytic leukemia. Blood 89:2015–2023
Lens D, Matutes E, Catovsky D (2000) Frequent deletions at 11q23 and 13q14 in B cell prolymphocytic leukemia (B-PLL). Leukemia 14:427–430
Flatley E, Chen AI, Zhao X (2014) Aberrations of MYC are a common event in B-cell prolymphocytic leukemia. Am J Clin Pathol 142(3):347–354
Put N, Van Roosbroeck K, Konings P (2012) Chronic lymphocytic leukemia and prolymphocytic leukemia with MYC translocations: a subgroup with an aggressive disease course. Ann Hematol 91:863–873
Huh YO, Lin KI, Vega F (2008) MYC translocation in chronic lymphocytic leukaemia is associated with increased prolymphocytes and a poor prognosis. Br J Haematol 142:36–44
Kim B, Kim E, Lee ST, Cheong JW, Lyu CJ, Min YH et al (2020) Detection of recurrent, rare, and novel gene fusions in patients with acute leukemia using next-generation sequencing approaches. Hematol Oncol 38(1):82–88. https://doi.org/10.1002/hon.2709
Engvall M, Cahill N, Jonsson BI, Höglund M, Hallböök H, Cavelier L et al (2020) Detection of leukemia gene fusions by targeted RNA-sequencing in routine diagnostics. BMC Med Genomics 13:106. https://doi.org/10.1186/s12920-020-00739-4
Merchant S, Schlette E, Sanger W (2003) Mature B-cell leukemias with more than 55% prolymphocytes: report of 2 cases with Burkitt lymphoma-type chromosomal translocations involving c-myc. Arch Pathol Lab Med 127:305–309
Lens D, Coignet LJ, Brito-Babapulle V (1999) B cell prolymphocytic leukaemia (B-PLL) with complex karyotype and concurrent abnormalities of the p53 and c-MYC gene. Leukemia 13:873–876
Chow KU, Kim SZ, von Neuhoff N (2011) Clinical efficacy of immunochemotherapy with fludarabine, epirubicin and rituximab in the treatment for chronic lymphocytic leukaemia and prolymphocytic leukaemia. Eur J Haematol 87(5):426–433
Mourad YA, Taher A, Chehal A, Shamseddine A (2004) Successful treatment of B-cell prolymphocytic leukemia with monocloncal anti-CD20 antibody. Ann Hematol 83(5):319–321
Perz J, Topaly J, Fruehauf S (2002) Level of CD20-expression and efficacy of rituximab treatment in patients with resistant or relapsing B-cell prolymphocytic leukemia or B-cell chronic lymphocytic leukemia. Leuk Lymphoma 43(1):149–151
Tempescul A, Feuerbach J, Ianotto J-C (2009) A combination therapy with fludarabine, mitoxantrone and rituximab induces complete immunophenotypical remission in B-cell prolymphocytic leukaemia. Ann Hematol 88(1):85–88
Weide R, Pandorf A, Heymanns J, Koppler H (2004) Bendamustine/Mitoxantrone/Rituximab (BMR): a very effective, well toleroutpatient chemoimmunotherapy for relapsed and refractory CD20-positive indolent malignancies: final results of a pilot study. Leuk Lymphoma 45(12):2445–2449
Eyre TA, Fox CP, Shankara P. Idelalisib-Rituximab induces clinical remissions in patients with TP53 disrupted B cell prolymphocytic leukaemia. Br J Haematol. 2016.
Gordon MJ, Raess PW, Young K. Ibrutinib is an effective treatment for B-cell prolymphocytic leukaemia. Br J Haematol. 2016.
Chapiro E, Pramil E, Diop M (2019) Genetic characterization of B-cell prolymphocytic leukemia: a prognostic model involving MYC and TP53. Blood 134:1821–1831
Kay NE, Hanson CA (2019) B-cell prolymphocytic leukemia has 3 subsets. Blood 134:1777–1778
Hew J, Pham D, Matthews Hew T, Minocha V (2018) A novel treatment with obinutuzumab-chlorambucil in a patient with B-cell prolymphocytic leukemia: a case report and review of the literature. J Investig Med High Impact Case Rep 6:2324709618788674
George P, Brown A, Weinkove R (2020) B-cell prolymphocytic leukaemia with a t (4;14) FGFR3/IGH translocation: response to ibrutinib. Pathology 52(4):491–492. https://doi.org/10.1016/j.pathol.2020.03.005
Moore J, Baran AM, Meacham PJ, Evans AG, Barr PM (2020) Initial treatment of B-cell prolymphocytic leukemia with ibrutinib. Am J Hematol 95(5):E108–E110. https://doi.org/10.1002/ajh.25733
Jain A, Khunger JM, Prasad P, Chaudhry S, Sharma M, Gupta DK, Saluja S (2020) An illustrative case of B-cell prolymphocytic leukemia. Blood Res 55:175–189
Bell S, Lattanzio N, Braham J, Campdesuner V, Abdelal Q, Vartanov A et al (2021) An unusual case of prolymphocytic leukemia transformation in a patient with chronic lymphocytic leukemia. J Investig Med High Impact Case Rep. https://doi.org/10.1177/2324709621990767
Acknowledgements
All authors would like to thank patient family for participating in this report.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
MM, MM MAR, YY, JS, GL, NH, SAAEK, DM are shared in designing the research report, processing the samples, interpretation the patient data regarding the hematological disease and writing the case report and read and approved the final manuscript. All authors equally contributed.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The research methodology in the present work was approved by research ethical committee of Shandong University, and written informed consent was obtained from participant family enrolled in the case report prior to sample collection. Experimental procedures and sampling followed the international and national regulations in accordance with the Declaration of Helsinki.
Consent for publication
The authors grant the publisher permission to publish this work.
Competing interests
All authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Makongoro, M., Abu Rakhey, M.M.M., Yu, Y. et al. A new case of trisomy 5 with complex karyotype abnormalities in B-cell prolymphocytic leukemia: a case study. Egypt J Med Hum Genet 23, 39 (2022). https://doi.org/10.1186/s43042-022-00257-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-022-00257-1