
Abstract
Background
Src kinase, a nonreceptor protein-tyrosine kinase is composed of 11 members (in human) and is involved in a wide variety of essential functions required to sustain cellular homeostasis and survival.
Main body of the abstract
Deregulated activity of Src family kinase is related to malignant transformation. In 2001, Food and Drug Administration approved imatinib for the treatment of chronic myeloid leukemia followed by approval of various other inhibitors from this category as effective therapeutics for cancer patients. In the past decade, Src family kinase has been investigated for the treatment of diverse pathologies in addition to cancer. In this regard, we provide a systematic evaluation of Src kinase regarding its mechanistic role in cancer and other diseases. Here we comment on preclinical and clinical success of Src kinase inhibitors in cancer followed by diabetes, hypertension, tuberculosis, and inflammation.
Short conclusion
Studies focusing on the diversified role of Src kinase as potential therapeutical target for the development of medicinally active agents might produce significant advances in the management of not only various types of cancer but also other diseases which are in demand for potent and safe therapeutics.
Similar content being viewed by others

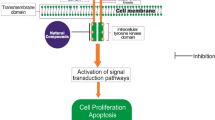
Background
The group of diseases in which the body cells grow and divide uncontrollably is collectively termed as cancer. Cancer is a diseased state in which the regulatory processes of the body like senescence and apoptosis are arrested. Cancer is the second leading cause of death globally, accounting for an estimated 9.6 million deaths (in 2018) as per World Health Organization (WHO) data. In 2016, 7.2 million cancer cases and 8.9 million deaths due to cancer were reported worldwide [1]. In 2016, all cancers together contributed to 5.0% of the total Disability-adjusted life years (DALYs) and 8.3% of the total deaths in India [2]. Cancerous cells lose their structure and function due to insufficient differentiation that results in cell mass formation in the affected area of the body. Cancer development occurs in three main phases namely initiation, promotion, and progression. Benign tumors can rarely become malignant, and the activities of some enzymes namely, hexokinase, phosphofructokinase, aldolase, enolase, and pyruvate kinase have been compared for benign and normal tissues [3]. Unlike other health threats contributing to the global burden of diseases, cancer represents a group of drastically different diseases that require unique and specific approaches for prevention, diagnosis, and treatment [1]. The United Nations Sustainable Development Goals target the reduction of premature mortality from non-communicable diseases, which includes cancer, to one-third by 2030 through prevention and treatment [4].
Various causes of cancer are possible, but all these causes eventually lead to the activation of proto-oncogenes. Classification of oncogenes can be done based on the functional and biochemical properties of protein products of proto-oncogenes. The groups in which oncogenes are classified are growth factors like platelet-derived growth factor (PDGF), growth factor receptors (ErbB, ErbB-2, Fms, Kit, Met, Ret, Ros, and Trk), signal transducers (non-receptor protein kinases and guanosine triphosphate binding proteins), transcription factors (ErbA, Ets, Fos, Jun, Myb, and c-Myc), and others, including programmed cell death regulators bcl-2 [5]. Src is a protein-coding gene and the protein encoded by this gene is protein kinases. Src, the proto-oncogene is useful for cytoskeletal formation via PI3K, MAPK, STAT-3, IL-8, and VEGF signaling pathways. Cellular Src (known as c-Src), was the first proto-oncogene to be identified [6]. The Nobel Prize in Physiology or Medicine, 1989 was awarded jointly to J. Michael Bishop and Harold E. Varmus for their discovery of the cellular origin of retroviral oncogenes [7]. In 1978, Raymond Erikson and coworker Marc Collette isolated the Src protein at the University of Colorado Medical Center [8].
Kinase, as defined by the National Cancer Institute, is an enzyme that acts by phosphorylation of other molecules like sugars or proteins. Phosphorylated proteins aid in signal transduction and is an important component of various signaling pathways and thus the knowledge of regulatory functions of kinases can be useful to identify more effective anticancer agents [9]. Active triphosphate forms of nucleoside analogues are used as anticancer agents as they can arrest elongation and/or inhibit enzymes essential for deoxyribonucleic acid (DNA) synthesis when incorporated into growing DNA strand [10]. Most of the protein kinases in normal physiology promote cell proliferation, differentiation, and migration, but when it is activated or overexpressed, they promote carcinogenesis. Kinase inhibitors are the most used categories of chemotherapy medicine. Unlike the extreme side effects of conventional chemotherapy, kinase inhibitors have high clinical efficacy with comparatively lesser toxic effects upon initial administration. The observed high frequency of acquired resistance in the case of kinase inhibitors suggests use of multi-targeted approaches that can rapidly eliminate the cancer cells by inhibiting multiple pathways. It helps to control the cancer cells by inhibition at multiple levels [11]. Over 30 anticancer kinase inhibitors are approved in the US, and many more are under development. The US FDA has approved a total of 52 small molecule protein kinase inhibitors as of 1st January 2020 [12]. The present manuscript aims to give an overview of kinase enzymes, their classification followed by importance of Src kinase as potential target for development of its inhibitors as anticancer therapeutics. The manuscript has also focused on the diversified role of Src kinase inhibitors in the development of therapeutics other than anticancer.
Classification of kinase enzymes
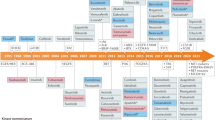
Depending on the type of substrate target, the kinase enzymes can be classified into protein kinase, lipid kinase, and carbohydrate kinase. Among all other kinase types, protein kinase is of utmost importance as the human genome can code over 500 of these kinases, and the phosphorylation of proteins by kinase enzymes regulates many cellular functions of the body. The genes that catalyze the phosphorylation of proteins are collectively termed as “kinome” [13]. Researchers have observed that among the reported 518 kinase genes of the human genome, 244 map cancers [14]. Among the 52 US FDA-approved drugs in the period 2015–2019, only 11 could inhibit protein-serine/threonine protein kinases; two are directed against MEK1/2, 11 block non-receptor protein-tyrosine kinases, and 28 target receptor protein-tyrosine kinases [12].
Classification of protein kinase as per the KinBase resource divides the kinases into two major groups that are conventional eukaryotic protein kinases (ePKs) and atypical protein kinases (aPKs) (Fig. 1) [15]. The aPKs have a dissimilar sequence from ePKs, but experimentally, they show protein kinase activities [16].

Classification of protein kinases
According to amino acid residues of proteins that are phosphorylated, the protein kinases are classified into the following families [17]:
-
1.
Serine/threonine protein kinases: cyclin-dependent kinase, mitogen-activated protein kinase (MAPK), protein kinase D, nattokinase, DNA-dependent protein kinase, Aurora protein kinases, pancreatic kininogenase
-
2.
Tyrosine-specific protein kinases: non-receptor tyrosine protein kinases (NRTKs), receptor tyrosine kinases (RTKs), nuclear tyrosine protein kinase
-
3.
Histidine-specific protein kinases: branch chain α-ketoacid dehydrogenase kinase (BCKDHK), pyruvate dehydrogenase kinase (PDHK)
-
4.
Tryptophan kinase
-
5.
Aspartyl/glutamyl protein kinase
Types of tyrosine protein kinase
Out of the 518 kinase genes in human, 90 have been identified as protein tyrosine kinases among which 58 are of receptor tyrosine kinase type, and 32 are of non-receptor tyrosine kinase type [18]. Tyrosine protein kinase is further divided into subcategories as receptor tyrosine kinases (RTKs), non-receptor tyrosine kinases (NRTKs), and nuclear protein tyrosine kinase which are detailed in the coming sections.
RTKs
RTKs have a large extracellular ligand-binding domain, some part in the trans membrane region, and an intracellular catalytic domain that selectively binds to and phosphorylates the substrate proteins after dimerization of receptors. The transmission of signals is through PI3K/AKT/mTOR, RAS/RAF/MEK/ERK, PLCγ/PKC, and other signaling pathways. RTKs include epidermal growth factor receptor (EGFR), insulin receptor (IR), fibroblast growth factor receptor (FGFR), vascular endothelial growth factor receptor (VEGFR), nerve growth factor (NGF), platelet-derived growth factor receptors (PDGFR), and many more. EGFR helps to regulate cell growth and differentiation. EGFR overexpression is reported in various epithelial tumor cells. Insulin receptor (IR) enhances the proliferation and inhibits apoptosis in breast cancer, cervical cancer, colon cancer, and lung cancer. FGFR is involved in the development of new blood vessels, and VEGFR plays a crucial role in the proliferation, migration, and vascularization of endothelial cells. PDGFR regulates cell growth and development and is mainly present in fibroblasts and smooth muscle cells, but it also affects the kidney, testis, and brain [18].
Non-receptor tyrosine protein kinases (NRTKs)
NRTKs is a group of nine kinase enzymes namely, ABL, ACK, CSK, FAK, FES, JAK, SRC, SYK, and TEC. Unlike RTKs, they do not have any intrinsic catalytic domain; instead, the agonist-induced dimerization will change the conformation of the intracellular domain in a way that its affinity for cytosolic protein kinase increases. The signals are transduced via free-moving proteins that can bind and are phosphorylated by the protein kinase.
The ubiquitously expressed Abelson (ABL) kinase family includes ABL1 and ABL2 proteins. The translocation of the breakpoint cluster region (BCR) sequences with the c-ABL tyrosine kinase to produce the BCR-ABL chimeric gene which has an enhanced tyrosine kinase activity. Activated Cdc42 kinases (ACKs) have subtypes ACK1/TNK2, ACK2, DACK, TNK1, ARK1, DPR2, and KOS1. Feline sarcoma (FES) kinase family inhibitors show relief in human lymphoid malignancies, colorectal cancer, and small-cell lung cancer. JAK family comprises four members namely, JAK1, JAK2, JAK3, and TYK2. Spleen tyrosine kinase (SYK) is one of the important classes of soluble cytosolic NRPKs [19].
Nuclear tyrosine protein kinase
The anticancer mechanism of nuclear tyrosine protein kinase is not well known. It is proposed that after ligand binding, the activated RTKs get internalized and then translocated to the endosomal compartments. It may bind to specific DNA sequences and produce proteins required for regulating activities of the target cells. Nuclear signaling is observed in the holoreceptor tyrosine kinase [20]. Some of the approved drugs acting on respective targets are mentioned in Table 1 [6, 20].
Structure of Src family kinases
General structure of Src family kinase
Src family kinases (SFKs) are a family of cytoplasmic tyrosine kinases. SFKs have a unique structure as shown in Fig. 2 that includes an N-terminal region with a 14-carbon myristoyl group, two Src homology domains (SH2 and SH3), a catalytic tyrosine-protein kinase domain (SH1), and a short C-terminal tail [21]. The SH1 domain has catalytic function, while the SH2 and SH3 domains have non-catalytic regulatory property, but all three domains are important for signal transduction [22, 23].

Organization of human Src kinase
Active sites of enzyme
The c-Src kinase catalytic domain has a peptide-binding lobe and an ATP-binding lobe. The substrate-docking site is constituted of180 residues and a peptide-binding lobe, but all of them are not involved in docking. Among them, six residues (Ser-273, Arg-279, Ser-280, Arg-281, Arg-283, and Phe-382) are significant determinants of the substrate-docking site as per the Ala scanning tests. Lee et al. verified the importance of these six residues using two mutants, one containing double mutations (DM) of Arg281Ala and Arg283Ala, and the other containing quadruple mutations (QM) of Ser280Ala, Arg281Ala, Arg283Ala, and Phe382Ala [24]. Cellular protein (c-Src) is different from the viral Src (v-Src), and the region containing Tyr527 is absent in the latter. Tyr527 phosphorylation is inhibitory to kinase activity, so the enzyme is constitutively active without it.
Biological activity of Src tyrosine kinase enzyme
Src tyrosine kinase enzymes are involved in the signaling pathways that control a diverse spectrum of biological activities that include gene transcription, immune response, cell adhesion, cell cycle progression, cell differentiation, apoptosis, movement, transformation, proliferation, and other essential cellular functions [25].
Activation of enzyme
Mechanism
As shown in Fig. 3, the activation segment present on the Src tyrosine kinase enzyme is Y419 that is Tyr419, which promotes kinase activity [21]. SFKs are activated by a series of steps, and the relative molecular mechanism varies upon cell types and extracellular cues. The process of activation starts from the activation of receptors, adaptors, or effectors and their interaction with the SH2/3 domains of inactive SFKs to open the closed conformation. The activated SFKs relocate to appropriate intracellular locations and the exposed pTyr-527 dephosphorylates to stabilize the active conformation [26]. Intermolecular autophosphorylation of the Src protein occurs at the activation loop Tyr419 [21]. Reorientation of the helix C is necessary to alleviate constraints and to establish a functional kinase [27].

Crystal structure of human tyrosine protein kinase Src
Enzyme activators
On activation, Src tyrosine kinase enzymes can phosphorylate various proteins like mitogen-activated protein kinase (MAPK), p38, and extracellular signal-regulated kinase (ERK). MAPKs regulate inflammation, cell development, cell differentiation, and cell senescence. While p38 is associated with permeability, survival, and migration of endothelial cells, ERK is involved in the proliferation and inflammation of endothelial cells. 4-Hydroxynonenal (4-HNE) is a major end product of lipid peroxidation. The HNE-enhanced activation of MAPKs/AP-1 signaling pathway and increased COX-2 expression is modulated by Src [28].
Role of Src kinases in cancer
Overall effects observed through clinical trials
Studies have proved that Src is not directly involved in tumor formation and is present in various pathways that promote cell division and growth. So, anti-Src monotherapy will not be an efficacious anticancer agent. But Src kinase inhibition may play an important auxiliary role in various cancer treatments due to its involvement in primary cell functions [21].
Mechanism of therapeutic activity
Phosphorylation at a specific tyrosine by CSK inactivates c-Src kinase, whereas autophosphorylation is essential for its activation process. Src kinase is one of the targets for anticancer drugs because it is involved in the signaling pathways [29]. The migration and proliferation of many normal cells are inhibited by contact inhibition, but cancer cells are insensitive to contact inhibition of growth [30].
Metastasis
Metastasis is the spread of cancer cells from the primary site to new areas of the body via the lymph system or circulatory system. The cancer cells can penetrate in new capillaries, and thus cancer cells can enter the circulatory system and begin the metastatic process [30]. Inhibition of c-Src kinase in Ewing’s sarcoma cells decreases migration and metastasis [31].
Invasion
The property of cancer cells to migrate into different tissue compartments from the primary site of growth, survive, and proliferate under these conditions is known as invasive growth [30]. By invasion, cells can reach the neighboring tissues via direct penetrations, and circulation is not involved in this type of tumor cell movement.
Angiogenesis
Angiogenesis supports tumor growth as new cells require new blood vessels for the supply of oxygen and nutrients to the proliferating tumor cells. Angiogenesis occurs in response to growth factors that stimulate endothelial cells of blood vessels [30].
Apoptosis
Apoptosis is a programmed cell death pathway of multicellular organisms in which the cells die by activation of enzymes capable of degrading the cell’s genetic material and proteins. Many cancer cells fail to undergo apoptosis, and these cells contribute substantially towards tumor development [30]. Overexpression of Src kinase reduces apoptosis as observed in U2OS, MG63 osteosarcoma cells, and Ewing’s sarcoma cells [31].
Src kinase inhibitors in clinical trials [32]
Various drugs from the category of kinase inhibitors have entered different phases of clinical trials. Among them, some are novel drugs, while most of the drugs are repurposed and are approved for some or the other type of cancer. Inhibition of Src kinase may also block immune responses that make the body prone to infections [33]. Dasatinib targets p-Src (Y527) in triple negative breast neoplasms and Tyr 419 phosphorylation of Src in mesenchymal stromal cell [34, 35]. Tyrosine kinase inhibitors for the MET gene are classified as type I (type Ia and type Ib), type II, and type III. Type I MET inhibitors bind to the active form of MET’s ATP-pocket. If we consider Crisotinib, it interacts with the Y1230 residue, the hinge field, and the G1163 solvent front. Capmatinib, Tepotinib, and Savolitinib have a strong connection with the Y1230 residue and the hinge, but no interaction with G1163. Bozitinib and TPX-022, the two new type I inhibitors are currently in clinical trials. Type II inhibitors, such as Cabozantinib, Merestinib, and Glesatinib, extend to a hydrophobic back pocket to bind the ATP-pocket in the inactive state. Both type I and type II inhibitors are ATP-competitive. Tivantinib is a type III inhibitor that binds to allosteric sites and is non-ATP-competitive [36]. TPX-0046 is RET/SRC inhibitor that has shown activity towards different RET-mutations including the G810R solvent front mutation [37]. In Table 2, the Src kinase inhibitors present in the clinical trial phases are included.
Src kinase inhibitors
In the development of kinase inhibitory therapeutics, generating highly selective probes to interrogate protein kinase function is a challenge. To overcome the limitations, many strategies have been effectively proposed which includes extension of the traditional inhibitor design by appending functionality proposed to interact with the specific loop of Src kinase, combinatorial chemistry, and high-throughput screening. Many of the developed inhibitors are small molecules and these are detailed here.
Classification
Kinase inhibitors are classified into three classes based on their capacity to catalyze the phosphorylation of substrates that usually contain serine, threonine, or tyrosine residue. Type I kinase inhibitors are ATP competitors that mimic the purine ring of the adenine moiety of ATP. They bind to active sites and cause conformational changes on ATP-binding sites. They do not offer a high degree of selectivity to kinase target and hence may show cardiotoxic effects. Type II kinase inhibitors target the inactive conformation of kinases by interacting with their catalytic site and are more selective as compared with Type 1 kinase inhibitors. The allosteric kinase inhibitors bind on the allosteric binding site of the substrate other than the ATP binding site. Type III kinase inhibitors are highly selective. The substrate-directed kinase inhibitors show a reversible interaction outside the ATP binding region that is present in the kinase substrate-binding site. The Type IV kinase inhibitors are the ATP noncompetitive inhibitors that offer a higher degree of selectivity against targeted kinases. The covalent kinase inhibitors bind to the kinase active site covalently in irreversible manner. Type V kinase inhibitors target a catalytic nucleophile cysteine. The exposed cysteine side chain in the ATP site is the approachable target in developing a drug candidate [38]. The approved drugs of each class of kinase inhibitors are mentioned in Table 3.
QSAR modification
Quantitative structure–activity relationship (QSAR) models include the study of molecular descriptors. As a result of QSAR modifications, novel drug structures are derived that have better properties. QSAR modeling utilizes the information about the chemical structure and their chemical activity to select lead compounds on which further studies are done for drug discovery. Using this technique, numerous Src kinase inhibitors are discovered that are enlisted in Table 4 and Fig. 4.

Src-kinase inhibitors developed from QSAR modification
Various studies have been reported for use of QSAR for virtual screening and design of novel kinase inhibitors [42,43,44]. The mechanism of tyrosine kinase receptor (EGFR, FGFR4, PDGFRA, and VEGFR2) inhibition by pyrazolopyrimidine derivatives has been derived using BP-ANN, multi-QSAR and molecular docking approach. It led to investigation of eight bioactive compounds, and only one of them could bind to all the four receptors when compared with the known drugs, gefitinib and regorafenib [45]. In a recent study, Ancuceanu et al. [46] report development of over 350 QSAR classification models using a ChEMBL dataset of 1038 compounds. The prediction results were confirmed using online version of PASS and are found to be better than that expected from the high-throughput screening experiments.
Multi-target anti cancerous drugs
For optimizing cancer therapeutics, the new approach to anticancer drug discovery includes design and development of multi-target agents by performing multiple docking, use of common pharmacophores, and fragment-based design. Recent advancement in the multi-target anticancer agents lead to use of target combinations as a potential approach for the development of rational drugs for improved efficacy.
The possible target combination could be the following [39]:
-
1.
Multi-targeted agents targeting tyrosine kinase and inducing DNA damage
-
2.
Multi-targeted agents targeting EGFR and Src kinase
-
3.
Multi-targeted agents targeting thymidylate synthase (TS) and dihydrofolate reductase (DHFR)
-
4.
Multi-targeted agents targeting aromatase and steroid sulfatase
-
5.
Multi-targeted agents targeting kinesin spindle protein (KSP) and aurora-A kinase
-
6.
Multi-targeted agents targeting tyrosine kinase and microtubule
-
7.
Multi-targeted agents targeting estrogen receptor a (ERa) and vascular endothelial growth factor receptor-2 (VEGFR-2)
-
8.
Multi-targeted agents targeting histone deacetylase (HDAC) and other targets
Role of Src kinases in other diseases
The activity of Src is supervised by tyrosine phosphorylation at two sites (Tyr527 and Tyr418) with unlike effects. The phosphorylation of Tyr527 at the COOH-terminal regulatory region results in the inactivation of Src, while phosphorylation of Tyr418 at the catalytic region results in the activation of Src [47]. Src are the overall key regulators of biological activities required for life. Src kinase controls a wide range of cellular events such as cell growth, division, differentiation, survival, and programmed cell death. The Src-associated focused roles include immune responses, cell adhesion, migration, and endocytosis. Hyperactivation of Src kinase has been concerned in the etiology of human diseases including cancer [48, 49]. Src kinase inhibitors have been implicated and have demonstrated therapeutic potential for diseases other than cancer. Some of them are elaborated in this section.
Inflammation
Inflammation pathway encompasses chemical mediators that include nitric oxide, reactive oxygen species, prostaglandin E2 (PGE2), histamine, cytokines like TNF-α, and several interleukins [50]. Research has revealed the vital role of Src in macrophage-mediated inflammatory responses. A range of inflammatory diseases are closely associated with macrophage activation. In response to the stimulation by LPS and other pathogens, cells can produce multiple cytokines and chemokines, such as TNF-α, IL-1, and IL-6 [51]. HCK, FGR, and LYN are the major Src kinase in these cells. It revealed that LPS stimulation improved Hck gene expression in human peripheral blood monocyte-derived macrophages [52]. Additionally, a collaboration of LPS and IFN brought an expression of Hck and Lyn in murine bone marrow-derived macrophages [53]. Some research indicate that inflammatory stimuli in monocytes and macrophages could cause the emergence of Src kinase. Some studies prove that Src PTK activities are regulated during inflammatory responses where epithelial cells, smooth muscle cells, and fibroblasts play an important role. The reported evidence shows that lung epithelial cells may contribute effectively to inflammatory responses [54, 55]. These cells can produce inflammatory mediators such as monocyte chemoattractant protein IL-1, IL-6, and IL-8 [56]. Moreover, recent studies have shown that human lung epithelial cells formed long pentraxin PTX3, a novel exposed mediator of innate immunity and inflammatory responses, in response to TNF-α and IL-1b [57]. Some anthraquinones, such as anthraquinone-2-carboxylic acid, flavonoids like Scutellarein, show anti-inflammatory activity by suppressing the inflammatory nuclear factor NF-κB in the signaling pathway by blocking Src kinase [58, 59]. Olea europaea methanol extract and Celtis choseniana methanol extract show the anti-inflammatory effects primarily by targeting Src in the NF-κB signaling pathway [60, 61]. The catalytic activity of SFKs suppresses LPS-induced inflammatory responses [62]. Recently, several research labs have explored the opportunities to use Src inhibitors against inflammatory diseases (Table 5).
Hypertension
Hypertension is known to be a major risk factor for congestive heart failure (CHF), coronary artery disease, stroke, renal disease, and peripheral vascular disease. Currently available treatment for primary hypertension emphasizes on inhibition of vascular resistance by annoying peptide hormones responsible for vasoconstriction and includes Angiotensin-II (Ang II), catecholamines and calcium channels [69]. Ang II is a bioactive peptide and plays a crucial role in cardiovascular homeostasis and pathogenesis [70]. Earlier research studies revealed that Ang II encourages vasoconstriction via multiple cellular signaling pathways. It is responsible for activation of AT1 receptor to bind Gq/11 and Gi/o proteins. This leads to activation of phospholipase C (PLC) and thus increases the concentrations of Ca2+ in the cytosol. It leads to activation of Ca2+/calmodulin-dependent MLC kinase, MAPKs (ERK1/2, JNK, and p38 kinase), protein kinase C, and tyrosine kinases including SFK [71,72,73,74,75]. Several documents exposed that SKF activation is the initial event in Ang II-induced signal transduction, and SKF plays a vital role in Ang II-induced vascular responses like ERK1/2 activation via proliferation, contraction, and cell migration [76,77,78]. However, the role of SFK for arterial contractions and its contribution to Ang II-induced hypertension is currently unknown. Bo Qin et al. found that SKF inhibitors SU6656 notably lowered the level of systemic blood pressure in Ang II-treated mice, which is related to phosphorylation of the smooth muscle myosin light chain (MLC) in the mesenteric-resistant blood vessels [79]. Gp130 is a potential therapeutic target to improve heart regeneration after cardiac injury by activating Yap via Src kinase [80].
Skin aging
Sulfuretin suppresses UV-induced MMP-1 expression by occupying ATP binding sites and thus suppresses SFKs. Upregulation of MMP-1 is responsible for accelerated skin aging [81]. UVB and α-MSH stimulates melanogenesis which causes inhibited Src phosphorylation in G361 cells. This indicates that c-Src inhibition can regulate UV- and α-MSH-induced pigmentation. In G361 cells, Src inhibition-induced melanogenesis through the p38 MAPK and PKA signaling pathways is reported. Also, c-Src activity inhibitors stimulate muscle differentiation through p38 MAPK activation. While there is little to no evidence that Src inhibitors affect CREB phosphorylation, it is logical to believe that p38 activation will trigger it. CREB phosphorylation is known as a downstream signal of p38 during UV-induced melanogenesis. In melanocytes, the MC1R and c-Kit signaling pathways may be linked to c-Src signaling. In melanocytes, inhibition of Src kinase may increase melanogenesis through p38 and CREB signaling pathways [82].
Chronic kidney disease (CKD)
CKD is an emerging worldwide public health issue [83]. Existing therapeutics for the management of CKD are ineffective. Thus, recognizing and validating novel therapeutic targets is supreme to investigate potent and safe options. In the past decades, intensive studies for the characterization of diverse key signal pathways and mediators in the pathogenesis of CKD have been reported [84, 85]. So far, many factors were identified including transforming growth factor-b1 (TGF-b1), and Ang II as impressive fibrogenic mediators. However, the pharmacological constraint of these factors shows inadequate therapeutic effectiveness. Yan et al. [86] has established and revealed that Src kinase is activated in kidney fibroblasts in response to TGF-b1 or serum and the fibrotic kidney after unilateral ureteral obstructions. Blockade of Src with PP1 or quieting it by small interfering RNA (siRNA) blocks myofibroblastic activation of fibroblast (in vitro) and ameliorates renal fibrosis (in vivo) after unilateral ureteral obstructions. Inhibition of Src by PP1 seems to interrupt TGFb1/Smad3 and epidermal growth factor receptor (EGFR) signaling (Fig. 5). This study shows that Src kinase acts as an integrator of multiple fibrogenic signals triggered by the activation of numerous membrane receptors. Chen et al. [87] also defined activation of Ang II receptor can stimulate Src activation. It mediates continuous phosphorylation and TGF expression of EGFR. Thus, Src may be a novel target for therapeutic intervention in fibrotic CKD. In Table 6, we outline the role of Src kinase inhibitors in chronic renal diseases with their respective mechanism.

Outline of Src family kinase in renal disease. Damage in the kidney induced Src kinase activation that causes transactivation of EGFR. Transactivated EGFR stimulates the overproduction of TGF-1 and similarly Smad3 signaling, a pathway for activation of renal fibroblasts and accumulation in extracellular matrix proteins. Smad3 also plays a role in mediating Hck-induced renal fibrosis and STAT3 required for Src- and Fyn-stimulated profibrotic responses in the kidney
Diabetes
Defects in insulin secretion and action lead to diabetes mellitus, and it is characterized by hyperglycemia. Type 1 diabetes mellitus (T1DM) is caused by autoimmune destruction of pancreatic β cells. T cell infiltration escorted due to cytokine and reactive oxygen species production, ultimately leads to β cell dysfunction and apoptosis [94, 95]. Type 2 diabetes mellitus (T2DM) involves development of resistance by the target tissues to metabolic actions of insulin and dysfunction of pancreatic β cells [96]. In case of high glucose-induced diabetic complications, the role of Src kinase is not reported. Insulin receptor is an RTK, and some studies have evidenced its role in diabetes [97]. Adult mouse islets are known to express KIT in β cells [98]. The c-Kit receptor appears to play a regulatory role in glucose metabolism. In a mouse model, c-Kit point mutation causes alteration of ATP-binding domain of the c-Kit receptor tyrosine kinase, impaired fasting glucose, and impaired glucose tolerance [99]. These mice also had a 50% reduction in β cell mass when compared with control mice. Additionally, inhibition of Src family tyrosine kinase results in improved calcium-induced insulin secretion equally in rat pancreatic islets and INS-2 cells suggesting that Src kinases may have an inhibitory role in calcium-dependent insulin secretions [100]. Several clinical cases of T1DM and T2DM reversal during TKI management have been stated. In vivo and in vitro experimental studies have tried to establish the mechanism behind this effect. Inhibition of Abelson tyrosine kinase (c-Abl) results in β cell survival and improved insulin secretion. Although, platelet-derived growth factor receptor (PDGFR) and EGFR hindrance lead to enhanced insulin sensitivity. Moreover, inhibition of vascular endothelial growth factor receptor 2 (VEGFR2) diminishes the degree inflammation in islet cell (insulitis). Thus, targeting several PTKs may provide a novel approach for correcting the pathophysiological disturbances associated with diabetes.
Evaluation of Src inhibitors by in vivo experiments in diabetic models indicate presence of high glucose-Src-tumor necrosis factor-α-converting enzyme (TACE), heparin-binding epidermal growth factor receptor (EGFR), and other signaling pathways. It concentrates on the use of Src as a novel therapeutic target for diabetic neuropathy. In STZ-diabetic mice, PP2 can inhibit albuminuria and increase Src pTyr-416, as well as cause TACE activation, ERK and EGFR phosphorylation, glomerular collagen accumulation, and podocyte loss [90].
Epilepsy
Epilepsy is a central nervous system (neurological) chronic disorder in which brain activity becomes abnormal, causing recurrent seizures, unusual behavior and sensations, and sometimes loss of consciousness [101]. An epileptic brain can be characterized with axon and dendritic growth development, deviations in receptor compositions, synaptic development, and preservations. The inflammatory processes are controlled by cell signaling pathways [102,103,104,105,106]. Inhibition of these pathways have potential to prevent formation of epileptic circuitry and/or prevent the development of epilepsy after brain injury.
In chronic epilepsy, kinase signaling comprising of activated JAK-STAT, BDNF-TrkB, and PI3K-Akt-mTOR pathways have been demonstrated in animal and in vitro models [107,108,109,110,111,112,113,114]. Till date, a small percentage of kinases have been found to play a major role in epilepsy [115]. Several kinases play role in glia, neurons, and microglia [116], and there is a possibility that they may play a vital role in epileptogenesis and/or the development of epilepsy. Some kinase inhibitors are not always specific inhibitors and need to be experimentally determined [117,118,119]. Due to the availability of inhibitors, only a few kinases have been studied for preventing or altering epilepsy including FGFRs, VEGFRs, Flt, EGFR, Erbb receptors, IGF-1R, c-Met, cFMS, GM-CSFR, and PDGFRs as well as some neurotrophin receptors. All the receptor tyrosine kinase listed above and the phosphorylation changes have been discussed in the epileptic brain in the literature [120,121,122,123]. Total Src protein expression increases in symptomatic epileptic tissues when compared with the control group, but the expression of Src-pY416 in human symptomatic epileptic tissues was significantly decreased. Results of the study suggested that the decreased GluN2B phosphorylation in human symptomatic epileptic tissues may be regulated by the NRG1-ErbB4-Src signaling pathway [124].
Tuberculosis
Tuberculosis (TB) is an airborne infection that most often affects lungs and may spread to other body parts. TB is caused by Mycobacterium tuberculosis and is contagious. It has escorted to mankind throughout the history and never stopped to affect them. TB is still among the top 10 causes of death pandemic around the globe. Globally, a projected 1.7 billion (23%) of the world’s population is infected with Mycobacterium tuberculosis leading to more than 10 million cases with TB every year. According to the report of the WHO, around 1.5 million people died from TB. Yet, the wide duration of therapy and the emergence of multidrug-resistant tuberculosis (MDR-TB) have created a supreme need to discover more selective new anti-TB drugs for effective treatment. Chandra et al. reported that Src plays the central character in defining cellular responses simultaneous to infection with Mycobacterium tuberculosis. In response, they conclude signaling events upstream from transcriptional regulation of Src and downstream from activated Src (pY416) would hold the key to establish the mechanistic ability of the entire process [125]. Moreover, studies on Src inhibition have shown a significant effect on survival of H37Rv, MDR, and extremely drug-resistant (XDR) strains of Mycobacterium tuberculosis. In THP-1 macrophages, Src inhibition also plays a crucial role in reducing survival and can regulate TB infection in guinea pigs [126]. Thus, Src kinase inhibitors could be established into the host-directed anti-TB drugs.
Conclusions
Over the past few decades, role of Src kinases in biological system has been well elucidated. Like other members of the kinase family, Src kinases play an important role in cancer cell proliferation, angiogenesis, and chemoresistance. Thus, Src kinase inhibitors are focused as therapeutic agents for the treatment of metastatic disease. Preclinical and clinical data supports the potential utility of Src kinase inhibitors by designing anticancer therapeutics. Recent studies are going on to investigate their benefit as combination regimen. In addition, hyperactivation of Src has been evaluated as possible target for CKD, inflammation, diabetes, epilepsy, hypertension, and TB. Studies conclude signaling events arising from the transcriptional regulation of Src and downstream from activated Src would be the key towards establishment of Src kinase inhibitors as diversified therapeutics.
Future studies focusing on the diversified role of Src as potential therapeutical target for the development of medicinally active agents might produce significant advances in the management of not only various types of cancer but also other diseases which are in demand for potent and safe therapeutics.
Availability of data and materials
Not applicable.
Abbreviations
- 4-HNE:
-
4-Hydroxynoneal
- ABL:
-
Abelson kinase
- ACK:
-
Activated Cdc42 kinase
- ALI:
-
Acute lung injury
- ALK:
-
Anaplastic lymphoma kinase
- ALL:
-
Acute lymphoblastic leukemia
- AML:
-
Acute myeloid leukemia
- aPKs:
-
Atypical protein kinases
- BCKDHK:
-
Branch chain α-ketoacid dehydrogenase kinase
- BTK:
-
Bruton tyrosine kinase
- CDK:
-
Cyclin dependent kinase
- CHF:
-
Congestive heart failure
- CKD:
-
Chronic kidney disease
- CML:
-
Chronic myeloid leukemia
- Col4:
-
Collagen 4
- COX:
-
Cyclooxygenase
- CSK:
-
C-terminal Src kinase
- DALYs:
-
Disability-adjusted life years
- DHFR:
-
Dihydrofolate reductase
- DM:
-
Double mutation
- DNA:
-
Deoxyribonucleic acid
- EGFR:
-
Epidermal growth factor receptor
- EMT:
-
Epithelial–mesenchymal transition
- ePKs:
-
Eukaryotic protein kinases
- Era:
-
Estrogen receptor a
- ERK:
-
Extracellular signal-regulated kinase
- FAK:
-
Focal adhesion kinase
- FES:
-
Feline sarcoma
- FGFR:
-
Fibroblast growth factor receptor
- FLT3:
-
FMS-like tyrosine kinase 3
- GBM:
-
Glomerular base membrane
- HDAC:
-
Histone deacetylase
- IR:
-
Insulin receptor
- JAK:
-
Janus kinase
- KSP:
-
Kinesin spindle protein
- MAPK:
-
Mitogen-activated protein kinase
- MDR-TB:
-
Multidrug-resistant tuberculosis
- MEK:
-
Mitogen-activated kinase
- MLC:
-
Myosin light chain
- NGF:
-
Nerve growth factor
- NRTK:
-
Non-receptor tyrosine protein kinases
- PDGF:
-
Platelet-derived growth factor
- PDGFR:
-
Platelet-derived growth factor receptors
- PDHK:
-
Pyruvate dehydrogenase kinase
- PGE2:
-
Prostaglandin E2
- PI3K:
-
Phosphoinositide 3-kinase
- PLC:
-
Phospholipase C
- PP:
-
4-Amino-5-(4-methylphenyl)-7-(t-butyl) pyrazolo[3,4-d]-pyrimidine
- QM:
-
Quadruple mutations
- QSAR:
-
Quantitative structure–activity relationship
- RTK:
-
Receptor tyrosine kinases
- SFK:
-
Src kinase family
- SH:
-
Src homology domain
- siRNA:
-
Small interfering RNA
- SYK:
-
Spleen tyrosine kinase
- TACE:
-
Tumor necrosis factor-converting enzyme
- TACE:
-
Tumor necrosis factor-α-converting enzyme
- TGF-b1:
-
Transforming growth factor-b1
- TS:
-
Thymidylate synthase
- US FDA:
-
United States Food and Drug Administration
- VEGFR:
-
Vascular endothelial growth factor receptor
- WHO:
-
World Health Organization
- XDR:
-
Extremely drug-resistant
References
Global Burden of Disease Cancer Collaboration (2018) Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 29 cancer groups, 1990 to 2016: a systematic analysis for the Global Burden of Disease Study. JAMA Oncol. 4(11):1553–1568. https://doi.org/10.1001/jamaoncol.2018.2706
India State-Level Disease Burden Initiative Cancer Collaborators (2018) The burden of cancers and their variations across the states of India: the Global Burden of Disease Study 1990-2016. Lancet Oncol. 19(10):1289–1306
Hennipman A, Smits J, van Oirschot B, van Houwelingen JC, Rijksen G, Neyt JP, Unnik JAV, Staal GE (1987) Glycolytic enzymes in breast cancer, benign breast disease and normal breast tissue. Tumor Biol. 8(5):251–263. https://doi.org/10.1159/000217529
Cao B, Bray F, Ilbawi A, Soerjomataram I (2018) Effect on longevity of one-third reduction in premature mortality from non-communicable diseases by 2030: a global analysis of the Sustainable Development Goal health target. Lancet Glob Health. 6(12):e1288–e1296. https://doi.org/10.1016/S2214-109X(18)30411-X
Pierotti MA, Sozzi G, Croce CM (2003) Discovery and identification of oncogenes. In: Kufe DW, Pollock RE, Weichselbaum RR et al (eds) Holland-Frei Cancer Medicine, 6th edn. BC Decker, Hamilton
Kannaiyan R, Mahadevan D (2018) A comprehensive review of protein kinase inhibitors for cancer therapy. Expert Rev Anticancer Ther. 18(12):1249–1270. https://doi.org/10.1080/14737140.2018.1527688
Hunter T, Sefton BM (1980) Transforming gene product of Rous sarcoma virus phosphorylates tyrosine. Proc Natl Acad Sci USA. 77(3):1311–1315. https://doi.org/10.1073/pnas.77.3.1311
Collett MS, Erikson RL (1978) Protein kinase activity associated with the avian sarcoma virus src gene product. Proc Natl Acad Sci USA. 75(4):2021–2024. https://doi.org/10.1073/pnas.75.4.2021
Ardito F, Giuliani M, Perrone D, Troiano G, Lo ML (2017) The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int J Mol Medi. 40(2):271–280. https://doi.org/10.3892/ijmm.2017.3036
Galmarini CM, Mackey JR, Dumontet C (2002) Nucleoside analogues and nucleobases in cancer treatment. Lancet Oncol. 3(7):415–424. https://doi.org/10.1016/S1470-2045(02)00788-X
Ho VWT, Tan HY, Wang N, Feng Y. Cancer management by tyrosine kinase inhibitors: efficacy, limitation, and future strategies. Tyrosine Kinases as Druggable Targets in Cancer. 2019. https://www.intechopen.com/books/tyrosine-kinases-as-druggable-targets-in-cancer/cancer-management-by-tyrosine-kinase-inhibitors-efficacy-limitation-and-future-strategies Accessed 10 Dec 2020.
Roskoski R (2020) Properties of FDA-approved small molecule protein kinase inhibitors: a 2020 update. Pharmacol Res. 152:104609. https://doi.org/10.1016/j.phrs.2019.104609
Modi V, Dunbrack RL (2019) A structurally - validated multiple sequence alignment of 497 human protein kinase domains. Sci Rep. 9(1):19790. https://doi.org/10.1038/s41598-019-56499-4
Kerr S (2013) Drug discovery through enzyme inhibition. In: Lemke TL, Williams DA, Roche VF, Zito SW (eds) Foye’s principles of medicinal chemistry, 7th edn. Lippincott Williams & Wilkins, Philadelphia, p 303
Uniport Database. https://www.uniprot.org/docs/pkinfam Accessed 2 Dec 2020.
Richardson CJ, Gao Q, Mitsopoulous C, Zvelebil M, Pearl LH, Pearl FM (2009) MoKCa database-mutations of kinases in cancer. Nucleic Acids Res. 37(suppl_1):D824–D831. https://doi.org/10.1093/nar/gkn832
Cusabio. https://www.cusabio.com/c-16643.html Accessed 10 Dec 2020.
Jiao Q, Bi L, Ren Y, Song S, Wang Q, Wang YS (2018) Advances in studies of tyrosine kinase inhibitors and their acquired resistance. Mol Cancer. 17(1):36. https://doi.org/10.1186/s12943-018-0801-5
Azevedo A, Silva S, Rueff J. Non-receptor tyrosine kinases role and significance in hematological malignancies. Tyrosine Kinases as Druggable Targets in Cancer. 2019. https://www.intechopen.com/books/tyrosine-kinases-as-druggable-targets-in-cancer/non-receptor-tyrosine-kinases-role-and-significance-in-hematological-malignancies Accessed 10 Dec 2020.
Song S, Rosen KM, Corfas G (2013) Biological function of nuclear receptor tyrosine kinase action. Cold Spring Harb Perspect Biol. 5(7):a009001
Roskoski R Jr (2015) Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol. Res. 94:9–25. https://doi.org/10.1016/j.phrs.2015.01.003
El-Rashedy AA, El-Din AAM (2018) Drug design of Src kinase inhibitor: an overview. J Innovat Pharm Biol Sci. 5(1):51–59
RCSB Protein Data Bank. http://www.rcsb.org/ Accessed 10 Dec 2020.
Lee S, Lin X, Nam NH, Parang K, Sun G (2003) Determination of the substrate-docking site of protein tyrosine kinase C-terminal Src kinase. Proc Nat Acad Sci. 100(25):14707–14712. https://doi.org/10.1073/pnas.2534493100
Uniport Database. https://www.uniprot.org/uniprot/P12931 Accessed 2 Dec 2020.
Okada M (2012) Regulation of the SRC family kinases by Csk. Int J Biol Sci. 8(10):1385–1397. https://doi.org/10.7150/ijbs.5141
Wenqing XU, Harrisont SC, Eckt MJ (1997) Three-dimensional structure of the tyrosine kinase c-Src. Nature. 385(6617):595–602
Jang EJ, Jeong HO, Park D, Kim DH, Choi YJ, Chung KW, Park MH, Yu BP, Chung HY (2015) Src tyrosine kinase activation by 4-hydroxynonenal upregulates p38, ERK/AP-1 signaling and COX-2 expression in YPEN-1 cells. Plos One. 10(10):e0129244. https://doi.org/10.1371/journal.pone.0129244
Cui Z, Chen S, Wang Y, Gao C, Chen Y, Tan C, Jiang Y (2017) Design, synthesis and evaluation of azaacridine derivatives as dual-target EGFR and Src kinase inhibitors for antitumor treatment. Eur J Med Chem. 136:372–381. https://doi.org/10.1016/j.ejmech.2017.05.006
Cooper GM (2000) The cell: a molecular approach, 2nd edn. Sinauer Associates, Sunderland
Chen Q, Zhou Z, Shan L, Zeng H, Hua Y, Cai Z (2015) The importance of Src signaling in sarcoma (Review). Oncol Lett. 10(1):17–22. https://doi.org/10.3892/ol.2015.3184
Hana A, Leeb J, Leea M, Leec SY, Shina EJ, Songa YR, Leea KM, Leec KW, Lima TG (2019) Sulfuretin, a natural Src family kinases inhibitor for suppressing solar UV induced skin aging. J Funct Foods. 52:442–449. https://doi.org/10.1016/j.jff.2018.11.032
Chen J, Elfiky A, Han M, Chen C, Saif MW (2014) The role of Src in colon cancer and its therapeutic implications. Clin Colorectal Cancer. 13(1):5–13. https://doi.org/10.1016/j.clcc.2013.10.003
Canonici A, Browne AL, Fanning KP, Roche S, Conlon NT, O’Neill F, Meiller J, Cremona M, Morgan C, Hennessy BT, Eustace AJ, Solca F, O’Donovan N, Crown J (2020) Combined targeting EGFR and SRC as a potential novel therapeutic approach for the treatment of triple negative breast cancer. Ther Adv Med Oncol. 12:1–16
Boufker HI, Lagneaux L, Najar M, Piccart M, Ghanem G, Body JJ, Journé F (2010) The Src inhibitor dasatinib accelerates the differentiation of human bone marrow-derived mesenchymal stromal cells into osteoblasts. BMC Cancer. 10(1):298. https://doi.org/10.1186/1471-2407-10-298
Hong L, Zhang J, Heymach JV, Le X (2021) Current and future treatment options for MET exon 14 skipping alterations in non-small cell lung cancer. Ther Adv Med Oncol. 15(13):1758835921992976
Belli C, Anand S, Gainor JF, Penault-Llorca F, Subbiah V, Drilon A, Andrè F, Curigliano G (2020) Progresses toward precision medicine in RET-altered solid tumors. Clin Cancer Res 26(23):6102–6111
Bhullar KS, Lagarón NO, McGowan EM, Parmar I, Jha A, Hubbard BP, Rupasinghe HPV (2018) Kinase-targeted cancer therapies: progress, challenges and future directions. Mol Cancer. 17(1):48. https://doi.org/10.1186/s12943-018-0804-2
Fu R, Sun Y, Sheng W, Liao D (2017) Designing multi-targeted agents: an emerging anticancer drug discovery paradigm. Eur J Med Chem. 136:195–211. https://doi.org/10.1016/j.ejmech.2017.05.016
Sun D, Zhao Y, Zhang S, Zhang L, Liu B, Ouyang L (2020) Dual-target kinase drug design: current strategies and future directions in cancer therapy. Eur J Med Chem. 15(188):112025
Musumeci F, Fallacara AL, Brullo C, Grossi G, Botta L, Calandro P, Chiariello M, Kissova M, Crespan E, Maga G, Schenone S (2017) Identification of new pyrrolo[2,3-d]pyrimidines as Src tyrosine kinase inhibitors in vitro active against glioblastoma. Eur J Med Chem. 15(127):369–378
Anwer Z, Gupta SP (2011) A QSAR study on a series of indolin-2-ones acting as non-receptor Src tyrosine kinase inhibitors. Lett Drug Design Discov. 8(10):918–925. https://doi.org/10.2174/157018011797655250
Zhang P, Jie H, Liu J, Zhang XY, Zhang W, Liu M, Wang Y, Wang YF, Huang W, Liu Z (2020) Studies on substituted thienopyridine carbonitriles as Src inhibitors using a comprehensive in silico method. Ind J Pharm Sci. 82:270–281
Patil VM, Gupta SP, Masand N (2017) Quantitative structure-activity relationship studies: understanding the mechanism of tyrosine kinase inhibition. Curr Enzyme Inhi. 13:139–159
Bahmani A, Tanzadehpanah H, Hosseinpour Moghadam N, Saidijam M. Introducing a pyrazolopyrimidine as a multi-tyrosine kinase inhibitor, using multi-QSAR and docking methods. Mol Divers. 2020. https://doi.org/10.1007/s11030-020-10080-8. Epub ahead of print.
Ancuceanu R, Tamba B, Stoicescu CS, Dinu M (2019) Use of QSAR global models and molecular docking for developing new inhibitors of c-Src tyrosine kinase. Int J Mol Sci. 21(1):19. https://doi.org/10.3390/ijms21010019
Yeatman TJ (2004) A renaissance for SRC. Nat Rev Cancer. 4(6):470–480. https://doi.org/10.1038/nrc1366
Hunter T (2009) Tyrosine phosphorylation: thirty years and counting. Curr Opin Cell Biol. 21(2):140–146. https://doi.org/10.1016/j.ceb.2009.01.028
Patil VM, Masand N (2020) Role of kinases in antiviral therapeutics in A closer look at kinase inhibition. Edited by Alex Tompson. NOVA Publishers, USA
Del Donno M, Bittesnich D, Chetta A, Olivieri D, Lopez-Vidriero MT (2000) The effect of inflammation on mucociliary clearance in asthma: an overview. Chest. 118(4):1142–1149. https://doi.org/10.1378/chest.118.4.1142
Cohen J (2002) The immunopathogenesis of sepsis. Nature. 420(6917):885–891. https://doi.org/10.1038/nature01326
Ziegler SF, Wilson CB, Perlmutter RM (1988) Augmented expression of a myeloid-specific protein tyrosine kinase gene (hck) after macrophage activation. J Exp Med. 168(5):1801–1810. https://doi.org/10.1084/jem.168.5.1801
Boulet I, Ralph S, Stanley E, Lock P, Dunn AR, Green SP, Phillips WA (1992) Lipopolysaccharide-and interferon-gamma-induced expression of hck and lyn tyrosine kinases in murine bone marrow-derived macrophages. Oncogene. 7(4):703–710
Liu M. Alveolar epithelium in host defence: cytokine production. In Sepsis and Organ Dysfunction, Springer Milano. 2002;37-50, doi: https://doi.org/10.1007/978-88-470-2213-3_2.
Simon RH, Paine R (1995) 3rd. Participation of pulmonary alveolar epithelial cells in lung inflammation. J Lab Clin Med. 126(2):108–118
Kany S, Vollrath JT, Relja B (2019) Cytokines in inflammatory disease. Int J Mol Sci. 20(23):6008. https://doi.org/10.3390/ijms20236008
Han B, Mura M, Andrade CF, Okutani D, Lodyga M, dos Santos CC, Keshavjee S, Matthay M, Liu M (2005) TNFalpha-induced long pentraxin PTX3 expression in human lung epithelial cells via JNK. J Immunol. 175(12):8303–8311. https://doi.org/10.4049/jimmunol.175.12.8303
Park JG, Kim SC, Kim YH, Yang WS, Kim Y, Hong S, Kim KH, Yoo BC, Kim SH, Kim JH, Cho JY (2016) Anti-inflammatory and antinociceptive activities of anthraquinone-2-carboxylic acid. Mediators Inflamm. 2016:1903849
Sung NY, Kim MY, Cho JY (2015) Scutellarein reduces inflammatory responses by inhibiting Src kinase activity. Korean J Physiol Pharmacol. 19(5):441–449. https://doi.org/10.4196/kjpp.2015.19.5.441
Song C, Hong YH, Park JG, Kim HG, Jeong D, Oh J, Sung GH, Hossain MA, Taamalli A, Kim JH, Cho JY (2019) Suppression of Src and Syk in the NF-κB signaling pathway by Olea europaea methanol extract is leading to its anti-inflammatory effects. J Ethnopharmacol. 235:38–46. https://doi.org/10.1016/j.jep.2019.01.024
Kim HG, Choi S, Lee J, Hong YH, Jeong D, Yoon K, Yoon DH, Sung GH, Lee S, Hong S, Yi YS, Kim JH, Cho JY (2018) Src is a prime target inhibited by Celtis choseniana methanol extract in its anti-inflammatory action. Evid Based Complement Alternat Med. 2018:3909038
Mitchell J, Kim SJ, Seelmann A, Veit B, Shepard B, Im E, Rhee SH (2018) Src family kinase tyrosine phosphorylates Toll-like receptor 4 to dissociate MyD88 and Mal/Tirap, suppressing LPS-induced inflammatory responses. Biochem Pharmacol. 147:119–127. https://doi.org/10.1016/j.bcp.2017.11.015
Khadaroo RG, He R, Parodo J, Powers KA, Marshall JC, Kapus A, Rotstein OD (2004) The role of the Src family of tyrosine kinases after oxidant-induced lung injury in vivo. Surgery. 136(2):483–488. https://doi.org/10.1016/j.surg.2004.05.029
Severgnini M, Takahashi S, Tu P, Perides G, Homer RJ, Jhung JW, Bhavsar D, Cochran BH, Simon AR (2005) Inhibition of the Src and Jak kinases protects against lipopolysaccharide-induced acute lung injury. Am J Respir Crit Care Med. 171(8):858–867. https://doi.org/10.1164/rccm.200407-981OC
Kusaka G, Ishikawa M, Nanda A, Granger DN, Zhang JH (2004) Signaling pathways for early brain injury after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 24(8):916–925. https://doi.org/10.1097/01.WCB.0000125886.48838.7E
Lennmyr F, Ericsson A, Gerwins P, Akterin S, Ahlström H, Terént A (2004) Src family kinase-inhibitor PP2 reduces focal ischemic brain injury. Acta Neurol Scand. 110(3):175–179. https://doi.org/10.1111/j.1600-0404.2004.00306.x
Weis S, Shintani S, Weber A, Kirchmair R, Wood M, Cravens A, McSharry H, Iwakura A, Yoon YS, Himes N, Burstein D (2004) Src blockade stabilizes a Flk/cadherin complex, reducing edema and tissue injury following myocardial infarction. J Clin Invest. 113(6):885–894. https://doi.org/10.1172/JCI200420702
Paul R, Zhang ZG, Eliceiri BP, Jiang Q, Boccia AD, Zhang RL, Chopp M, Cheresh DA (2001) Src deficiency or blockade of Src activity in mice provides cerebral protection following stroke. Nat Med. 7(2):222–227. https://doi.org/10.1038/84675
Sever PS, Messerli FH (2011) Hypertension management 2011: optimal combination therapy. Eur Heart J. 32(20):2499–2506. https://doi.org/10.1093/eurheartj/ehr177
Schiffrin EL, Park JB, Intengan HD, Touyz RM (2000) Correction of arterial structure and endothelial dysfunction in human essential hypertension by the angiotensin receptor antagonist losartan. Circulation. 101(14):1653–1659. https://doi.org/10.1161/01.CIR.101.14.1653
Garrido AM, Griendling KK (2009) NADPH oxidases and angiotensin II receptor signaling. Mol Cell Endocrinol. 302(2):148–158. https://doi.org/10.1016/j.mce.2008.11.003
Higuchi S, Ohtsu H, Suzuki H, Shirai H, Frank GD, Eguchi S (2007) Angiotensin II signal transduction through the AT1 receptor: novel insights into mechanisms and pathophysiology. Clin Sci. 112(8):417–428. https://doi.org/10.1042/CS20060342
Tang DD, Anfinogenova Y (2008) Physiologic properties and regulation of the actin cytoskeleton in vascular smooth muscle. J Cardiovasc Pharmacol Ther. 13(2):130–140. https://doi.org/10.1177/1074248407313737
Cat AND, Touyz RM (2011) Cell signaling of angiotensin II on vascular tone: novel mechanisms. Curr Hypertens Rep. 13(2):122–128
Mehta PK, Griendling KK (2007) Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 292(1):C82–C97. https://doi.org/10.1152/ajpcell.00287.2006
Abe K, Nakashima H, Ishida M, Miho N, Sawano M, Soe NN, Kurabayashi M, Chayama K, Yoshizumi M, Ishida T (2008) Angiotensin II−induced osteopontin expression in vascular smooth muscle cells involves G q/11, Ras, ERK, Src and Ets-1. Hypertens Res. 31(5):987–998. https://doi.org/10.1291/hypres.31.987
Touyz RM, Wu XH, He G, Park JB, Chen X, Vacher J, Rajapurohitam V, Schiffrin EL (2001) Role of c-Src in the regulation of vascular contraction and Ca2+ signaling by angiotensin II in human vascular smooth muscle cells. J Hypertens. 19(3):441–449. https://doi.org/10.1097/00004872-200103000-00012
Mugabe BE, Yaghini FA, Song CY, Buharalioglu CK, Waters CM, Malik KU (2010) Angiotensin II-induced migration of vascular smooth muscle cells is mediated by p38 mitogen-activated protein kinase-activated c-Src through spleen tyrosine kinase and epidermal growth factor receptor transactivation. J Pharmacol Exp Ther. 332(1):116–124. https://doi.org/10.1124/jpet.109.157552
Qin B, Zhou J (2015) Src family kinases (SFK) mediate angiotensin II-induced myosin light chain phosphorylation and hypertension. Plos One. 10(5):0127891
Li Y, Feng J, Song S, Li H, Yang H, Zhou B, Li Y, Yue Z, Lian H, Liu L, Hu S, Nie Y (2020) gp130 Controls cardiomyocyte proliferation and heart regeneration. Circulation. 142(10):967–982. https://doi.org/10.1161/CIRCULATIONAHA.119.044484
Ge MM, Zhou YQ, Tian XB, Manyande A, Tian YK, Ye DW, Yang H (2020) Src-family protein tyrosine kinases: a promising target for treating chronic pain. Biomed Pharmacother 125:110017. https://doi.org/10.1016/j.biopha.2020.110017
Ku KE, Choi N, Oh SH, Kim WS, Suh W, Sung JH (2019) Src inhibition induces melanogenesis in human G361 cells. Mol Med Rep. 19(4):3061–3070. https://doi.org/10.3892/mmr.2019.9958
Glassock RJ, Warnock DG, Delanaye P (2017) The global burden of chronic kidney disease: estimates, variability and pitfalls. Nat Rev Nephrol. 13(2):104–114. https://doi.org/10.1038/nrneph.2016.163
Liu Y (2011) Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol. 7(12):684–696. https://doi.org/10.1038/nrneph.2011.149
Zeisberg M, Neilson EG (2010) Mechanisms of tubulointerstitial fibrosis. J Am Soc Nephrol. 21(11):1819–1834. https://doi.org/10.1681/ASN.2010080793
Yan Y, Ma L, Zhou X, Ponnusamy M, Tang J, Zhuang MA, Tolbert E, Bayliss G, Bai J, Zhuang S (2016) Src inhibition blocks renal interstitial fibroblast activation and ameliorates renal fibrosis. Kidney Int. 89(1):68–81. https://doi.org/10.1038/ki.2015.293
Chen J, Chen JK, Nagai K, Plieth D, Tan M, Lee TC, Threadgill DW, Neilson EG, Harris RC (2012) EGFR signaling promotes TGFβ-dependent renal fibrosis. J Am Soc Nephrol. 23(2):215–224. https://doi.org/10.1681/ASN.2011070645
Mima A, Abe H, Nagai K, Arai H, Matsubara T, Araki M, Torikoshi K, Tominaga T, Iehara N, Fukatsu A, Kita T (2011) Activation of Src mediates PDGF-induced Smad1 phosphorylation and contributes to the progression of glomerulosclerosis in glomerulonephritis. Plos one. 6(3):17929
Mima A, Matsubara T, Arai H, Abe H, Nagai K, Kanamori H, Sumi E, Takahashi T, Iehara N, Fukatsu A, Kita T (2006) Angiotensin II-dependent Src and Smad1 signaling pathway is crucial for the development of diabetic nephropathy. Lab Invest. 86(9):927–939. https://doi.org/10.1038/labinvest.3700445
Taniguchi K, Xia L, Goldberg HJ, Lee KW, Shah A, Stavar L, Masson EA, Momen A, Shikatani EA, John R, Husain M (2013) Inhibition of Src kinase blocks high glucose–induced EGFR transactivation and collagen synthesis in mesangial cells and prevents diabetic nephropathy in mice. Diabetes. 62(11):3874–3886. https://doi.org/10.2337/db12-1010
Wu H, Shi Y, Deng X, Su Y, Du C, Wei J, Ren Y, Wu M, Hou Y, Duan H (2015) Inhibition of c-Src/p38 MAPK pathway ameliorates renal tubular epithelial cells apoptosis in db/db mice. Mol Cell Endocrinol. 417:27–35. https://doi.org/10.1016/j.mce.2015.09.008
Elliott J, Zheleznova NN, Wilson PD (2011) c-Src inactivation reduces renal epithelial cell-matrix adhesion, proliferation, and cyst formation. Am J Physiol Cell Physiol. 301(2):C522–C529. https://doi.org/10.1152/ajpcell.00163.2010
Sweeney WE Jr, von Vigier RO, Frost P, Avner ED (2008) Src inhibition ameliorates polycystic kidney disease. J Am Soc Nephrol. 19(7):1331–1341. https://doi.org/10.1681/ASN.2007060665
Morgan NG, Leete P, Foulis AK, Richardson SJ (2014) Islet inflammation in human type 1 diabetes mellitus. IUBMB life. 66(11):723–734. https://doi.org/10.1002/iub.1330
Crèvecoeur I, Rondas D, Mathieu C, Overbergh L (2015) The beta-cell in type 1 diabetes: What have we learned from proteomic studies? Proteomics-Clin Appl. 9(7-8):755–766. https://doi.org/10.1002/prca.201400135
Tsatsoulis A, Mantzaris MD, Bellou S, Andrikoula M (2013) Insulin resistance: an adaptive mechanism becomes maladaptive in the current environment-an evolutionary perspective. Metab. 62(5):622–633. https://doi.org/10.1016/j.metabol.2012.11.004
Oakie A, Wang R (2018) β-Cell receptor tyrosine kinases in controlling insulin secretion and exocytotic machinery: c-kit and insulin receptor. Endocrinology. 159(11):3813–3821. https://doi.org/10.1210/en.2018-00716
Rachdi L, El Ghazi L, Bernex F, Panthier JJ, Czernichow P, Scharfmann R (2001) Expression of the receptor tyrosine kinase KIT in mature β-cells and in the pancreas in development. Diabetes. 50(9):2021–2028. https://doi.org/10.2337/diabetes.50.9.2021
Krishnamurthy M, Ayazi F, Li J, Lyttle AW, Woods M, Wu Y, Yee SP, Wang R (2007) c-Kit in early onset of diabetes: a morphological and functional analysis of pancreatic β-cells in c-Kit Wv mutant mice. Endocrinology. 148(11):5520–5530. https://doi.org/10.1210/en.2007-0387
Cheng H, Straub SG, Sharp GW (2007) Inhibitory role of Src family tyrosine kinases on Ca2+-dependent insulin release. Am J Physiol Endocrinol Metab. 292(3):E845–E852. https://doi.org/10.1152/ajpendo.00103.2006
Cheke RS, Firke SD, Patil RR, Bari SB. ISATIN: new hope against convulsion. Central Nervous System Agents in Medicinal Chemistry (Formerly Current Medicinal Chemistry-Central Nervous System Agents). 2018;18(2):76-101, doi: https://doi.org/10.2174/1871524917666171113124112.
Goldberg EM, Coulter DA (2013) Mechanisms of epileptogenesis: a convergence on neural circuit dysfunction. Nat Rev Neurosci. 14(5):337–349. https://doi.org/10.1038/nrn3482
Pitkänen A, Lukasiuk K (2009) Molecular and cellular basis of epileptogenesis in symptomatic epilepsy. Epilepsy Behav. 14(1):16–25. https://doi.org/10.1016/j.yebeh.2008.09.023
Amato S, Liu X, Zheng B, Cantley L, Rakic P, Man HY (2011) AMP-activated protein kinase regulates neuronal polarization by interfering with PI 3-kinase localization. Science. 332(6026):247–251. https://doi.org/10.1126/science.1201678
Chan CB, Liu X, Pradoldej S, Hao C, An J, Yepes M, Luo HR, Ye K (2011) Phosphoinositide 3-kinase enhancer regulates neuronal dendritogenesis and survival in neocortex. J Neurosci. 31(22):8083–8092. https://doi.org/10.1523/JNEUROSCI.1129-11.2011
Oliva AA, Atkins CM, Copenagle L, Banker GA (2006) Activated c-Jun N-terminal kinase is required for axon formation. J Neurosci. 26(37):9462–9470. https://doi.org/10.1523/JNEUROSCI.2625-06.2006
Berdichevsky Y, Dryer AM, Saponjian Y, Mahoney MM, Pimentel CA, Lucini CA, Usenovic M, Staley KJ (2013) PI3K-Akt signaling activates mTOR-mediated epileptogenesis in organotypic hippocampal culture model of post-traumatic epilepsy. J Neurosci. 33(21):9056–9067. https://doi.org/10.1523/JNEUROSCI.3870-12.2013
Aungst S, England PM, Thompson SM (2013) Critical role of trkB receptors in reactive axonal sprouting and hyperexcitability after axonal injury. J Neurophysiol. 109(3):813–824. https://doi.org/10.1152/jn.00869.2012
Scharfman HE (2005) Brain-derived neurotrophic factor and epilepsy—a missing link. Epilepsy Curr. 5(3):83–88. https://doi.org/10.1111/j.1535-7511.2005.05312.x
Grabenstatter HL, Del Angel YC, Carlsen J, Wempe MF, White AM, Cogswell M, Russek SJ, Brooks-Kayal AR (2014) The effect of STAT3 inhibition on status epilepticus and subsequent spontaneous seizures in the pilocarpine model of acquired epilepsy. Neurobiol Dis. 62:73–85. https://doi.org/10.1016/j.nbd.2013.09.003
Zeng LH, Rensing NR, Wong M (2009) The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J Neurosci. 29(21):6964–6972. https://doi.org/10.1523/JNEUROSCI.0066-09.2009
Liu G, Gu B, He XP, Joshi RB, Wackerle HD, Rodriguiz RM, Wetsel WC, McNamara JO (2013) Transient inhibition of TrkB kinase after status epilepticus prevents development of temporal lobe epilepsy. Neuron. 79(1):31–38. https://doi.org/10.1016/j.neuron.2013.04.027
Dinocourt C, Gallagher SE, Thompson SM (2006) Injury-induced axonal sprouting in the hippocampus is initiated by activation of trkB receptors. Eu J Neurosci. 24(7):1857–1866. https://doi.org/10.1111/j.1460-9568.2006.05067.x
Buckmaster PS, Ingram EA, Wen X (2009) Inhibition of the mammalian target of rapamycin signaling pathway suppresses dentate granule cell axon sprouting in a rodent model of temporal lobe epilepsy. J Neurosci. 29(25):8259–8269. https://doi.org/10.1523/JNEUROSCI.4179-08.2009
Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S (2002) The protein kinase complement of the human genome. Science. 298(5600):1912–1934. https://doi.org/10.1126/science.1075762
Martin KJ, Arthur JSC (2012) Selective kinase inhibitors as tools for neuroscience research. Neuropharmacology. 63(7):1227–1237. https://doi.org/10.1016/j.neuropharm.2012.07.024
Gao Y, Davies SP, Augustin M, Woodward A, Patel UA, Kovelman R, Harvey KJ (2013) A broad activity screen in support of a chemogenomic map for kinase signalling research and drug discovery. Biochem J 451(2):313–328. https://doi.org/10.1042/BJ20121418
Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, Mclauchlan H, Klevernic I, Arthur JSC, Alessi DR, Cohen P (2007) The selectivity of protein kinase inhibitors: a further update. Biochem J. 408(3):297–315. https://doi.org/10.1042/BJ20070797
Anastassiadis T, Deacon SW, Devarajan K, Ma H, Peterson JR (2011) Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat Biotechnol. 29(11):1039–1045. https://doi.org/10.1038/nbt.2017
Liu B, Chen H, Johns TG, Neufeld AH (2006) Epidermal growth factor receptor activation: an upstream signal for transition of quiescent astrocytes into reactive astrocytes after neural injury. J Neurosci. 26(28):7532–7540. https://doi.org/10.1523/JNEUROSCI.1004-06.2006
Nagayama T, Nagayama M, Kohara S, Kamiguchi H, Shibuya M, Katoh Y, Itoh J, Shinohara Y (2004) Post-ischemic delayed expression of hepatocyte growth factor and c-Met in mouse brain following focal cerebral ischemia. Brain Res. 999(2):155–166. https://doi.org/10.1016/j.brainres.2003.11.052
Sköld MK, Gertten CV, Sandbergnordqvist AC, Mathiesen T, Holmin S (2005) VEGF and VEGF receptor expression after experimental brain contusion in rat. J Neurotrauma. 22(3):353–367. https://doi.org/10.1089/neu.2005.22.353
Castañeda-Cabral JL, Beas-Zárate C, Rocha-Arrieta LL, Orozco-Suárez SA, Alonso-Vanegas M, Guevara-Guzmán R, Ureña-Guerrero ME (2019) Increased protein expression of VEGF-A, VEGF-B, VEGF-C and their receptors in the temporal neocortex of pharmacoresistant temporal lobe epilepsy patients. J Neuroimmunol. 328:68–72. https://doi.org/10.1016/j.jneuroim.2018.12.007
Zhu JM, Li KX, Cao SX, Chen XJ, Shen CJ, Zhang Y, Geng HY, Chen BQ, Lian H, Zhang JM, Li XM (2017) Increased NRG1-ErbB4 signaling in human symptomatic epilepsy. Sci Rep 7(1):141
Karim AF, Chandra P, Chopra A, Siddiqui Z, Bhaskar A, Singh A, Kumar D (2011) Express path analysis identifies a tyrosine kinase Src-centric network regulating divergent host responses to Mycobacterium tuberculosis infection. J Biol Chem. 286(46):40307–40319. https://doi.org/10.1074/jbc.M111.266239
Chandra P, Rajmani RS, Verma G, Bhavesh NS, Kumar D (2016) Targeting drug-sensitive and-resistant strains of Mycobacterium tuberculosis by inhibition of Src family kinases lowers disease burden and pathology. Msphere. 1:2
Acknowledgments
Authors acknowledge the financial support provided by Dr. A. P. J. Abdul Kalam Technical University, Lucknow (AKTU-VRPS) and facilities provided at KIET Group of Institutions, Ghaziabad, India for completing the work.
Funding
One of the authors (VMP) has received funding from Dr. A. P. J. Abdul Kalam Technical University, Lucknow, India under AKTU-Vishweshwarya Research Promotion Scheme funding (reference number is AKTU/Dean-PGSR/PhD/2019/5738) to perform the work.
Author information
Authors and Affiliations
Contributions
PN collected and analyzed data. PN and RSC have written the manuscript. VMP has developed the concept and supervised. The authors read and approved the final the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
Authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Negi, P., Cheke, R.S. & Patil, V.M. Recent advances in pharmacological diversification of Src family kinase inhibitors. Egypt J Med Hum Genet 22, 52 (2021). https://doi.org/10.1186/s43042-021-00172-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-021-00172-x