
Abstract
Fungi in the class Leotiomycetes are ecologically diverse, including mycorrhizas, endophytes of roots and leaves, plant pathogens, aquatic and aero-aquatic hyphomycetes, mammalian pathogens, and saprobes. These fungi are commonly detected in cultures from diseased tissue and from environmental DNA extracts. The identification of specimens from such character-poor samples increasingly relies on DNA sequencing. However, the current classification of Leotiomycetes is still largely based on morphologically defined taxa, especially at higher taxonomic levels. Consequently, the formal Leotiomycetes classification is frequently poorly congruent with the relationships suggested by DNA sequencing studies. Previous class-wide phylogenies of Leotiomycetes have been based on ribosomal DNA markers, with most of the published multi-gene studies being focussed on particular genera or families. In this paper we collate data available from specimens representing both sexual and asexual morphs from across the genetic breadth of the class, with a focus on generic type species, to present a phylogeny based on up to 15 concatenated genes across 279 specimens. Included in the dataset are genes that were extracted from 72 of the genomes available for the class, including 10 new genomes released with this study. To test the statistical support for the deepest branches in the phylogeny, an additional phylogeny based on 3156 genes from 51 selected genomes is also presented. To fill some of the taxonomic gaps in the 15-gene phylogeny, we further present an ITS gene tree, particularly targeting ex-type specimens of generic type species. A small number of novel taxa are proposed: Marthamycetales ord. nov., and Drepanopezizaceae and Mniaeciaceae fams. nov. The formal taxonomic changes are limited in part because of the ad hoc nature of taxon and specimen selection, based purely on the availability of data. The phylogeny constitutes a framework for enabling future taxonomically targeted studies using deliberate specimen selection. Such studies will ideally include designation of epitypes for the type species of those genera for which DNA is not able to be extracted from the original type specimen, and consideration of morphological characters whenever genetically defined clades are recognized as formal taxa within a classification.
Similar content being viewed by others

INTRODUCTION
The class Leotiomycetes was erected when the superclass Leotiomyceta was split into seven classes by Eriksson and Winka (1997). It is recognized as one of the most diverse classes in the subphylum Pezizomycotina, which in turn is one of the most diverse groups within Ascomycota (Berbee 2001). No recent estimates of the diversity of the entire Leotiomycetes have been made, but Hawksworth (2001) provided estimates for Erysiphales (10,000 spp.), a moderately well-studied order of Leotiomycetes, and Helotiales (70,000 spp.), a poorly studied order. Using these numbers as rough guides, we can expect a minimum of 80,000 species for the class. Current estimates put the number of published species in the class in the range 4407–5587, meaning that at most about 5–7% of the species diversity is currently known (Kirk et al. 2008; Baral 2016; Wijayawardene et al. 2017, 2018).
The class comprises non-lichenized ascomycetes, with those species that form a sexual morph historically referred to as “inoperculate discomycetes”. They are characterized by the production of an open ascoma (apothecium) and unitunicate asci generally opening by a pore (Eriksson 2005). In the last decade, this concept has changed mostly due to the contributions of molecular analyses in the systematics of the class. As currently conceived, the class is morphologically more diverse than Eriksson’s concept, including taxa with conspicuous or inconspicuous fruitbodies, usually apothecial, some opening late to expose the hymenium (pro- or mesohymenial phase) at maturity (e.g. Marthamycetaceae, Phacidiales, Rhytismatales), immersed in stromatic tissues or not, or with cleistothecial ascomata (e.g. Erysiphales). Some fungi now known to be members of Leotiomycetes have no known, or a rarely detected, sexual morph. These include vascular pathogens such as Cadophora, Phialocephala and Collophorina (Gams 2000; Damm et al. 2010; Day et al. 2012), the aquatic hyphomycetes (Baschien et al. 2013), mycorrhizal species, and root-inhabiting dark septate endophytes (Grünig et al. 2011).
The current classification of the class includes 11 orders, ca. 43 families, ca. 592 genera and ca. 4407 species, but these numbers vary depending on the taxonomic concepts applied by different authors, for example Baral (2016) versus Wijayawardene et al. (2017, 2018). Leotiomycetes also feature a remarkably high number of monotypic taxa: seven orders have only one family, two orders have only one species, 11 families have only one genus and ca. 231 genera have only one species (Baral 2016; Wijayawardene et al. 2017, 2018; Quijada 2018). The diversity is not equal across the class, Helotiales (ca. 2334 species) contains half of the described species, with three to five times more species than the second or the third most diverse orders, Erysiphales (ca. 747 spp.), and Rhytismatales (ca. 497 spp.). Helotiales is also the order in Leotiomycetes with the most genera placed as incertae sedis at the familial rank (ca. 90–151; Baral 2016, Wijayawardene et al. 2017, 2018, Quijada 2018). Currently, Helotiales includes 26 families, within which about 19–27% of the genera have an uncertain position at the family level (Baral 2016). Although the latest classifications of Leotiomycetes (op. cit.) have drastically changed the number of families and the placement of genera compared to earlier classifications (e.g. Lumbsch and Huhndorf 2010), the general concept of the order has remained the same for several decades and is now failing to reflect increasingly well-understood phylogenetic relationships.
An early class-wide phylogenetic analysis of Leotiomycetes, published as part of the AFTOL-1 project, was based on just the SSU, LSU and 5.8S rDNA regions (Wang et al. 2007). That study confirmed the placement of Geoglossaceae outside Leotiomycetes, the inclusion of Erysiphales and Rhytismatales in the class, and noted the polyphyly of Helotiales (Wang et al. 2007). Since then, other large phylogenies treating taxa within this class include Stenroos et al. (2010), Lantz et al. (2011), Han et al. (2014), Crous et al. (2014), Baral et al. (2015), and Pärtel et al. (2017). Lantz et al. (2011) demonstrated that some genera traditionally placed within Rhytismatales were related to other taxa placed in the polyphyletic Helotiales. Han et al. (2014) pointed out the extreme polyphyly of the family Hyaloscyphaceae, which constitutes at least 10 different clades within Leotiomycetes. The same year, Crous et al. (2014) circumscribed Phacidiaceae, previously considered a family in Helotiales, and accepted the order Phacidiales as a sister clade to Helotiales. A year later, Baral et al. (2016) suggested a new classification of Leotiomycetes. Baral (2016) provided more detail on thisnew classification, making many important changes at the family level, using both genetic and morphological evidence to establish several new families, to shift genera between families, and to revive several families that had fallen out of use. Baral (2016) accepted paraphyletic groups when strong morphological support was noted (e.g. Geoglossaceae was included in Leotiales, Tympanidaceae was included in Phacidiales). After this classification, three relevant publications appeared, Pärtel et al. (2017), Hernández-Restrepo et al. (2017), and Crous et al. (2017). In the first, evidence of the paraphyly of Helotiales, forming a clade that included Erysiphales and Cyttariales, was presented again, in a study that focused on the encoelioid taxa. The second established a new monotypic family and order for the genus Lauriomyces, and the third established two new families —Cochlearomycetacae and Neocrinulaceae —for two new, genetically isolated genera. Most of these phylogenies have a common problem, in that they are based on rDNA sequences which generally do not inform high-level relationships (Liu et al. 1999). Only four of these phylogenies included RPB2 sequences in combination with ribosomal DNA (rDNA) sequences (Stenroos et al. 2010; Han et al. 2014; Crous et al. 2014; Pärtel et al. 2017). The maximum number of gene regions used was six (Pärtel et al. 2017), and that is the only study to have used more than one protein-coding gene (EF1a, RPB1, and RPB2). Accounting for all of these phylogenies, only 32% of the genera included in Leotiomycetes have been incorporated at least once in a large phylogeny with more than three molecular markers (Quijada 2018). Although the families proposed by Baral (2016) are usually supported in these analyses, several of them are paraphyletic (e.g. Helotiaceae, Lachnaceae, Pezizellaceae, Rutstroemiaceae, Tympanidaceae), and in none of these phylogenies have the deeper, backbone relationships between families or orders been strongly supported. Several taxa remain as members of informal lineages (e.g. Bryoglossum, Stamnaria, Strossmayeria) sensu Baral (2016) or are placed as incertae sedis in Wijayawardene et al. (2017). Also, a high proportion of taxa have not yet been treated at all using molecular methods (Baral 2016; LoBuglio and Pfister 2010).
Most mycologists and users of fungal classifications, such as plant pathologists and mycorrhizal ecologists, now routinely identify their specimens using DNA sequences. Accurate morphological identifications depend on critical obseravtions and mycologists able to perform these are in rapid decline. Morphological characters can be misleading if incorrectly interpreted, being influenced by the methods used in morphological studies (Baral 1992) or the inherent morphological plasticity of many characters. Also, an increasing number of Leotiomycetes are known only from the character-poor asexual morph. DNA sequences provide an extremely high number of useable characters compared with the ones that we can find with morphology. Nevertheless, it is necessary to associate these sequences with reliable names because names link the sequences to historical, accumulated information on the biology, host range, distribution and pathogenic potential of the taxa detected (Crous et al. 2015). Reconciling morphology with phylogeny may not always be possible, but understanding molecular phylogenetic relationships as accurately as possible — and ideally reflecting them taxonomically in classifications — is important for understanding the role that fungi play in the ecosystems in which they have been detected. To enable this understanding, named fungarium specimens and cultures, especially type specimens, need to be linked with sequences (Truong et al. 2017).
Leotiomycetes has a worldwide distribution, and species have diverse ecological roles in soil, water or in the air, from the tropics to temperate, boreal or arctic-alpine, humid to arid ecosystems(O’Brien et al. 2005; Sieber 2007; Baral 2016). Taxa associated with fresh water include two families that characteristically form a sexual morph (Mitrulaceae and Vibrissaceae; Baral 2016), while many other genera from fresh-water are only known from an asexual morph (e.g. “aquatic hyphomycetes” in Raja et al. 2008, Gulis et al. 2012, Baschien et al. 2013). They have been found in many substrates in marine environments (Gnavi et al. 2014), including sponges in the Antarctic (Henríquez et al. 2014). Bryoglossum and Leotia form mycorrhizas, which have also been observed in some species of Mollisiaceae, Hyaloscyphaceae, and Myxotrichiaceae (Zijlstra et al. 2005; Kühdorf et al. 2015; Baral 2016). Endophytes have been found in several lineages of Dermateaceae, Gelatinodiscaceae, Helotiaceae, Phacidiaceae and Rhytismataceae (Baral 2016). Five families are exclusively plant pathogenic (Cyttariaceae, Erysiphaceae, Drepanopezizaceae, Medeolariaceae, and Sclerotiniaceae), one typically includes fungal parasites (Helicogonaceae), and some reportedly exhibit strong host specificity: the Stamnaria lineage (on Equisetum) and the Mniaecia lineage (on liverworts) (Baral 2016). Some members of the class are pathogenic to mammals, such as Pseudogymnascus destructans, which causes white-nose syndrome of bats (Blehert 2012). Despite this great diversity, the two predominant life-styles are saprobic and parasitic, with both strategies coexisting in the majority of families included in the class. Based on current sampling, the saprobic lifestyle appears to be the most common ecology in the class, more than half of the families having saprobic members growing on dead or decaying plant material (Baral 2016).
DNA-barcoding and high-throughput sequencing methods have changed the way fungal diversity in ecosystems can be measured. These new techniques allow the assessment of the fungal diversity in the environment that is independent of direct observation of fungal structures or cultivation of the fungi (Mitchell and Zuccaro 2006; Stewart 2012; Thomsen and Willerslev 2015). Purely molecular sampling methods have led to an increased number of sequences in general repositories, some identified but many unidentified, inspiring a debate as to whether sequences or eDNA should be allowed to serve as name-bearing types to describe and catalogue this hidden diversity (Hibbett et al. 2016; Hawksworth et al. 2018; Zamora et al. 2018; Hongsanan et al. 2018). The class Leotiomycetes provides a good example of how these techniques are revealing diversity, as members have been commonly found in permanently frozen soil (Lydolph et al. 2005), associated with roots (Hambleton and Sigler 2005; Toju et al. 2013; Hazard et al. 2014; Koizumi and Nara 2017), associated with bryophytes (Kauserud et al. 2008), on soil (Varma and Oelmüller 2007), in seawater (Henríquez et al. 2014), in air samples (Banchi et al. 2018), and elsewhere. Unfortunately, it is often impossible to associate these eDNA sequences with known species. In part this mismatch occurs because many taxa remain undiscovered and many named taxa lack DNA sequence data. Nevertheless, a classification of Leotiomycetes that better reflects molecular phylogeny would enable even the unknown taxa to be placed more reliably in a taxonomic framework, and through this placement their role in the ecosystems where they are detected can be better predicted (Wang et al. 2011).
In summary, the taxonomy and classification of Leotiomycetes remains unsettled. The current classification is still largely based on taxa defined by morphology and there is a disconnect between this morphology-based classification and molecular phylogenies. Because of its ecological diversity, the class has been studied by several distinct research communities, and this has resulted in the adoption of different classifications with different opinions about higher classification schemes. This problem needs to be solved, and the alternative schemes reconciled to develop one common classification. During the 11th International Mycological Congress in Puerto Rico (IMC11) in July 2018, a workshop on Leotiomycetes taxonomy was held, where 35 researchers were surveyed for their opinions on the current problems in the class: 80% of them agreed that the lack of specialist, general treatments, keys or sequences for common genera or species was a central issue; and 100% agreed that the main problem is polyphyly at different taxonomic levels (Quijada 2018). Here we use available data from published, taxonomically narrowly focused multi-gene studies, data extracted from published genomes, and some novel genome and sequence data, to provide a more robust class-wide phylogeny for Leotiomycetes. The aim is to provide a basis for taxon and specimen selection for future studies attempting to resolve outstanding issues associated with particular clades or subclades supported in the analyses presented here.
METHODS
Three sets of phylogenetic analyses were undertaken, one based on several thousand genes that were selected from 51 genomes, including the 10 new Leotiomycetes genomes released here (see below); a second based on up to 15 genes for a set of specimens selected to represent as widely as possible the genetic diversity across the class Leotiomycetes as understood from current rDNA-based class-wide phylogenies (e.g. Wang et al. 2007) and to include taxa known from both asexual and sexual morphs (e.g. Baschien et al. 2013); and a third, based on ITS sequences alone, which includes many taxa that were not represented in the other more data-rich analyses. Whenever possible, specimens were selected that represent the type species of a genus and where possible the type specimens themselves were used. If the only generic name available for a species is considered to be incorrect, it is cited in double quotes.
A complete list of the specimens and genomes sampled is given in Additional file 1: Table S1 and Additional file 2: Table S2.
Gene selection
The genes targeted for the 15-gene analysis were the ribosomal genes (SSU, 5.8S, LSU), and genes commonly used in recent multi-gene studies that include Leotiomycetes — β-tubulin, EF1a, MCM7, mtSSU, RPB1 and RPB2 (e.g. Baral et al. 2009; Chen et al. 2016; Crous et al. 2014; Han et al. 2014; Hosoya et al. 2010, 2011; Hustad and Miller 2011; Iturriaga et al. 2017; Lorch et al. 2013; Malloch et al. 2016; Pärtel et al. 2017; Sanoamuang et al. 2013; Schoch et al. 2009; Spatafora et al. 2006; Zhao et al. 2012).
Several additional genes were selected on the basis of a phylogenetic informativeness analysis using PhyDesign (Lόpez-Giráldez and Townsend 2011) to determine genes likely to be highly informative at deep levels within the phylogeny. An ultrametric tree was calculated for the taxa sampled in the single-copy orthologue dataset of Rokas et al. (2005) as modified by Taylor and Berbee (2006), using the methods of Taylor and Berbee (2006). Calibration points targeted divergence of major Pezizomycotina lineages, approximately 250–400 million years ago, based on the fungal, animal and plant calibrations of Taylor and Berbee (2006) (Additional file 3: Figure S1). Phylogenetic informativeness profiles were then calculated for gene fragments within the gene-rich taxon-poor sequence dataset of Taylor and Berbee (2006), and within the taxon-rich gene-poor dataset from the AFTOL-1 project (Schoch et al. 2009). Further consideration was dedicated to the 10 most informative genes for a timescale approximating early divergence within Leotiomycetes. These genes are listed, with their phylogenetic informativeness, along with potential primers for PCR amplification in Additional file 4: Table S3. Of these genes, RPB1 was already being targeted based on published multi-gene studies of the class. Preliminary attempts to amplify the other putatively highly informative genes were not consistently successful. However, several of these genes, RPA1, RPA2, RPC2, SF3B1, TFB4 and α-tubulin, were readily detected in the Leotiomycetes genomes we sampled (Additional file 1: Table S1). For those specimens that had genomes available, these genes were incorporated into the 15-gene analysis.
Newly generated DNA sequences
Newly generated Sanger sequences for SSU, ITS, LSU, β-tubulin, RPB1, RPB2, mtSSU, EF1a, and MCM7 were sourced from several laboratories, variously from cultures grown from ascospores discharged from apothecia, or from conidia from fresh water, cultures isolated from diseased plant tissue, and whole (single or multiple) apothecia. Sequences have been deposited in GenBank, or those that are unpublished or have been extracted from genomes, are available in the alignments provided in the Manaaki Whenua Datastore, https://doi.org/10.7931/T5YV-BE95. The voucher specimens from which the sequences were derived, GenBank accession numbers, and the researchers who generated the sequences are listed in Additional file 1: Table S1.
Genome sequencing and assembly
Genomic DNA for genome sequencing was isolated using the method of Schwessinger and McDonald (2017) and further purified using a QIAGEN Genomic-tip. Genomic DNAs were quantified using a fluorometer, and purity and integrity were assessed with a spectrophotometer and by agarose-gel electrophoresis. Sequencing was performed by Macrogen (South Korea) with an Illumina HiSeq2500 using a TruSeq Nano DNA kit with 100 bp paired-end reads. Multiplexing with 10 samples per lane resulted in 4.9–6.3 Gb per sample, with Q30 Phred quality score (99.9% base call accuracy) of 88–91%.
Short-read illumina sequence data was assembled using two different genome assembly tools: Platanus v. 1.2.4 (http://platanus.bio.titech.ac.jp/platanus-assembler/platanus-1-2-4), and the A5 Miseq Pipeline (Coil et al. 2015). For assembly with Platanus, sequence data was pre-processed using platanus_trim (http://platanus.bio.titech.ac.jp/pltanus_trim) followed by contig assembly, scaffolding, and gap closure using Platanus. The A5 Miseq pipeline used Trimmomatic (Bolger et al. 2014) for removal of adapter sequences and low-quality regions, followed by contig assembly using the IDBA-UD algorithm (Peng et al. 2012). Initial scaffolding, misassembly correction and final scaffolding was undertaken using SSPACE (Boetzer et al. 2010), bowtie (Langmead et al. 2009), samtools (Li et al. 2009), and BWA (Li and Durban 2009). QUAST (Gurevich et al. 2013) and BUSCO v3.0.2 (Simão et al. 2015; Waterhouse et al. 2017) were used to assess genome assembly completeness and quality. QUAST was run using the –eukaryote flag. BUSCO was run in genome mode with the Pezizomycotina odb9 database, of which Aspergillus nidulans is the default species.
Phylogenetic analyses
For the phylogeny based on 51 genomes, each genome was analyzed with BUSCO v3.0.2 using the Pezizomycotina odb9 database. This database contains 3156 putative single-copy reference genes which can be searched for within submitted genomes. The orthologous protein sequences identified using BUSCO were used for a phylogenetic analysis. Duplicate genes were removed from the phylogenetic analysis for those taxa where they were detected. Orthologous amino acid sequences were aligned separately using MUSCLE v. 3.8.31 (Edgar 2004). Following this alignment, gaps and phylogenetically uninformative positions were removed using Gblocks v. 0.91b (Castresana 2000; Talavera and Castresana 2007) with default settings. All aligned genes were concatenated into a supermatrix using Geneious v. 10 (Kearse et al. 2012), with sites of missing genes represented by N characters. A Bayesian Inference phylogeny was estimated using ExaBayes v. 1.5 (Aberer et al. 2014). Two independent runs were undertaken; following the recommended guidelines, analysis stopped after each had run for > 100,000 generations and the combined ESS values were > 100. Xylaria hypoxylon (JGI genome Xlyhyp1) and Neurospora crassa (JGI genome Neucr2) were used as outgroups.
For the phylogeny based on 15 genes, only 30 of the 279 specimens treated have all 15 genes available, but more than half have five or more genes (Additional file 5: Table S7). The sequences available for each gene were aligned using MAFFT (Katoh and Standley 2013) as implemented in Geneious 10. The ends were manually trimmed and introns were removed manually; all remaining data were then concatenated. Maximum likelihood analyses were run with IQ-TREE (Nguyen et al. 2015; Chernomor et al. 2016), using models selected by ModelFinder (Kalyaanamoorthy et al. 2017) for each partitioned gene; ultrafast bootstrap (BS) analysis with 1000 replicates estimated branch support in the ML tree (Hoang et al. 2018). Xylaria hypoxylon and Neurospora crassa were used as outgroups. The taxa and genes included in the alignment are provided in Additional file 1: Table S1 and the models used for each partition are in the Manaaki Whenua Landcare Research datastore, https://doi.org/10.7931/T5YV-BE95.
The ITS phylogeny includes 568 sequences. The methods for the analysis match those used for the 15-gene concatenated tree, but using the TIM2 + F + R9 model. Details of the taxa, specimens and GenBank sequence accessions are provided in Additional file 2: Table S2.
RESULTS
Results from the three phylogenetic analyses are provided as phylogenetic trees, one based on single copy genes across 51 genomes (Fig. 1), another based on up to 15 genes for 279 specimens across 68 genera (Figs. 2, 3, 4, 5 and 6), and a third using 568 ITS sequences (Additional file 6: Figure S2) representing 315 genera (including 80 generic type specimens). A comparison of the current family-level taxonomy with the taxonomic relationships suggested by our three sets of analyses is provided in Additional file 7: Table S6. The Supplementary Data, along with Nexus files of the 15-gene tree and the ITS tree, and the alignments on which they are based, can be downloaded from the Manaaki Whenua – Landcare Research datastore, see https://doi.org/10.7931/T5YV-BE95.

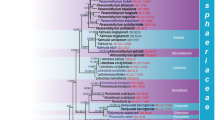
Phylogenetic tree based on a Bayesian analysis of 3156 concatenated orthologous single copy genes recognized using BUSCO from 49 selected Leotiomycetes genomes plus two outgroup genomes (Xylaria hypoxyolon and Neurospora crassa). Labels for the specimens sampled include the taxonomic name, voucher specimen where this data is available, source of the genome, and family level classification for that genus as accepted in Baral (2016). The large Helotiales clade is divided into several informal subclades that are discussed in the text

Summary tree from the ML analysis based on 15 concatenated sequences. Names for the collapsed family level clades are those accepted in this paper and generally the clades follow Baral (2016), the clades labelled Han 4, 7, 9 are those recognized in Han et al. (2014). Helotiales sensu Baral (2016) is divided into several informal clades based on bootstrap support in both this tree and Bayesian PP support in the genome-scale phylogeny (Fig. 1). The labels for taxa which are not included in one of the family-level clades include the voucher specimen from which the sequences were obtained, the type status of the specimen (whether it is the ex-type specimen of the type species for that genus, or whether it has been identified as the type species for that genus), the family in which it was placed by Baral (2016), if it was treated in that work, and the source of the genome data for those that have had their genome sequenced. Bootstrap values 90% or greater indicated, in a few cases lower bootstrap values for a few key branches are also indicated. Figures 3, 4, 5 and 6 provide detail of species included in each clade

Detail from Fig. 2. ML tree based on 15 concatenated sequences for specimens within the informal “mollisioid clade”, “discinelloid clade”, “hyaloscypheloid clade” and “erysiphoid clade” within Helotiales. Family level taxa indicated to the right are those accepted in this paper, ‘Han Clade 7’ is from Han et al. (2014). Thick branches have bootstrap values > 95%, lower bootstrap values for a few key branches are also indicated. The labels for taxa include the voucher specimen from which the sequences were obtained, the type status of the specimen (whether it is the ex-type specimen of the type species for that genus, or whether it has been identified as the type species for that genus), the family in which it was placed by Baral (2016), if it was treated in that work, and the source of the genome data for those that have had their genome sequenced. Some very long branches have been shortened for presentation; the Figs. 2, 3, 4, 5 and 6 nexus file in https://doi.org/10.7931/T5YV-BE95 has the original scale

Detail from Fig. 2. ML tree based on 15 concatenated sequences for specimens within the informal “helotioid clade” and “pezizelloid clade”, and the Stamanaria lineage within Helotiales. Family level taxa indicated to the right are those accepted in this paper, ‘Han Clade 4’ and ‘Han Clade 9’ are from Han et al. (2014). Thick branches have bootstrap values > 95%, lower bootstrap values for a few key branches are also indicated. The labels for taxa include the voucher specimen from which the sequences were obtained, the type status of the specimen (whether it is the ex-type specimen of the type species for that genus, or whether it has been identified as the type species for that genus), the family in which it was placed by Baral (2016), if it was treated in that work (*Vandijckellaceae was described in 2017), and the source of the genome data for those that have had their genome sequenced

Detail from Fig. 2. ML tree based on 15 concatenated sequences for specimens within the informal “sclerotinioid clade” and Dermateaceae within Helotiales. Family level taxa indicated to the right are those accepted in this paper. Thick branches have bootstrap values > 95%, lower bootstrap values for a few key branches are also indicated. The labels for taxa include the voucher specimen from which the sequences were obtained, the type status of the specimen (whether it is the ex-type specimen of the type species for that genus, or whether it has been identified as the type species for that genus), and the family in which it was placed by Baral (2016), if it was treated in that work, and the source of the genome data for those that have had their genome sequenced. Some very long branches have been shortened for presentation; the Figs. 2, 3, 4, 5 and 6 nexus file in https://doi.org/10.7931/T5YV-BE95 has the original scale

ML Detail from Fig. 2. tree based on 15 concatenated sequences for specimens within Marthamycetales, Leotiales, Thelebolales, Phacidiales, Rhytismatales and Chaetomellales. Family and order level taxa indicated to the right are those accepted in this paper. Thick branches have bootstrap values > 95%, lower bootstrap values for a few key branches are also indicated. The labels for taxa include the voucher specimen from which the sequences were obtained, the type status of the specimen (whether it is the ex-type specimen of the type species for that genus, or whether it has been identified as the type species for that genus), the family in which it was placed by Baral (2016), if it was treated in that work, and the source of the genome data for those that have had their genome sequenced. Some very long branches have been shortened for presentation; the Figs. 2, 3, 4, 5 and 6 nexus file in https://doi.org/10.7931/T5YV-BE95 has the original scale
Genome assembly and phylogeny
The 10 draft nuclear genomes have been deposited as NCBI Bioproject PRJNA487672. Data on genome size, coverage, GC content, number and size of contigs is provided in Additional file 8: Table S4. The A5 Miseq assembly pipeline generally provided greater coverage, higher N50 values, and a greater number of unique predicted genes compared with the Platanus assembler. Based on a BUSCO comparison, all but two of our genomes assembled using the A5 Miseq assembler provided a completeness greater than 98%; the other two provided 96.8 and 90.1% completeness (Additional file 9: Table S5).
The 10 newly generated genomes plus a further 41 genomes were selected for phylogenetic analysis. From all but one of these genomes, we were able to detect more than 80% of the 3156 genes in the BUSCO Pezizomycotina database, with sets of genes for most genomes being more than 98% complete (Additional file 9: Table S5). The only poorly sampled genome was the powdery mildew Golovinomyces cichoracearum (54.8% completeness), although a notably large number of duplicate genes were detected for Diplocarpon rosae.
The genome phylogeny had posterior probability support of > 0.95 for all branches (Fig. 1). The clades resolved are congruent with the family-level classification of Baral (2016). Strong support for deeper branches suggests that it is possible to identify a set of higher-level taxa within the current order Helotiales. Taxon sampling depth near the base of the tree is inadequate to draw any taxonomic conclusions for those taxa included that are in Leotiomycetes orders other than Helotiales.
15-gene phylogeny
Based on the 15-gene phylogeny, 258 species of Leotiomycetes were distributed across 31 formal and informal family-level clades as shown in the summary tree in Fig. 2. Placement of individual taxa within the clades is detailed in Figs. 3, 4, 5 and 6. Generally, the family-level relationships resolved in Figs. 2, 3, 4, 5 and 6 are congruent with the family level classification presented in Baral (2016). Exceptions are discussed below. Based on this phylogeny, a new order and two new families are introduced, along with the validation of an existing family name; these are formally treated in the Taxonomy section below. A series of informal clades are recognized within the large order Helotiales. The limits for these clades are based on support in the 15-gene phylogeny, as well as in the genome-scale phylogeny (Fig. 1).
Within the “mollisioid clade” of the Helotiales there is strong molecular phylogenetic support for a group comprising the familes Mollisiaceae, Vibrisseaceae, Loramycetaceae, the Strossmayeria lineage of Baral (2016) and the genus Chlorosplenium (incertae sedis in Baral 2016) (Figs. 2 and 3). Although within this group further strong molecular phylogenetic structure is recognisable, it remains a matter of debate whether or not the whole group should be included in the oldest family Mollisiaceae. Scattered across the subclades within the group are the type species of genera such as Acephala, Mollisia, Phialocephala and Vibrissea (Fig. 3). These generic names could be useful in naming some of these clades as part of a reworking of the generic classification within this group of families.
The Discinella-Pezoloma lineage proposed by Baral (2016) is strongly resolved. Along with the apothecial genera Pezoloma and Discinella, the family includes several aquatic genera known only as hyphomycete morphs — Articulospora, Gyoerffyella, Lemonniera, and Varicosporium (all sampled from specimens identified as the type species) (Fig. 3). Baral (2016) placed the Discinella-Pezoloma lineage with Gelatinodiscaceae, Helotiaceae and Roesleriaceae in his “Lineage B” (Helotiaceae s.lat.). In the present study, the Discinella-Pezoloma lineage and Gelatinodiscaceae are strongly supported as sister clades in the informal “discinelloid clade” of Helotiales, distant from Helotiaceae (incl. Roesleriaceae). Taxa in the Discinella-Pezoloma lineage differ from Gelatinodiscaceae by their paraphyses lacking vacuolar bodies, asci mostly with hemiamyloid apical rings of the Pezicula-type, the consistent absence of crystals, and asexual morphs being hyphomycetous with filiform or staurosporous conidia.
The “hyaloscyphoid clade” includes Hyaloscyphcaeae s. str., genera recognized by Han et al. (2014) as Clade 7, and the recently described mycorrhizal genus Cairneyella (Figs. 2 and 3). As discussed by Han et al. (2014), genera traditionally placed in Hyaloscyphaceae on the basis of morphology are polyphyletic. The type species of Hyaloscypha, H. vitreola, forms a strongly supported clade with root endophytic fungi in Meliniomyces and Rhizoscyphus (these latter two genera recently synonymised with Hyaloscypha, Fehrer et al. 2019), along with species in genera historically placed in Hyaloscyphaceae based on their ascoma morphology, Olla and Hyalopeziza. Also in this clade is a specimen that has had its genome sequenced and that was identified as Chalara longipes. Based on a comparison of SSU sequences, this specimen could be congeneric with a specimen identified as Chalara fusarioides (AF203463), the type species of the genus. Other genera traditionally placed in Hyaloscyphaceae are discussed below in the sections on Han Clade 9/Stamnaria lineage and the “pezizelloid clade”.
The “erysiphoid clade” includes the sister families Erysiphaceae and Arachnopezizaceae (Figs. 2 and 3). The powdery mildew family Erysiphaceae is traditionally treated as a separate order Erysiphales but is a molecular phylogenetic member of Helotiales as currently circumscribed.
The “helotioid clade” includes Helotiaceae and Lachnaceae (Figs. 2 and 4). Roesleriaceae, retained by Baral (2016) as a distinct family because of its unusual morphology, also belongs in Helotiaceae based on molecular phylogeny. The Helotiaceae clade has strong internal molecular phylogenetic structure (Fig. 4). “Hymenoscyphus” varicosporioides (genetically distinct from the type species of Hymenoscyphus and reportedly with a Varicosporium-like asexual morph later referred to the genus Tricladium; Sivichai et al. 2003) forms a well-resolved clade within Helotiaceae that in the 15-gene tree also includes the aquatic hyphomycete genera Mycofalcella and Tricladium. Two genera with reduced, cleistothecial ascomata currently classified as Pseudeurotiaceae — Connersia and Pleuroascus — belong in Helotiaceae, again in a well-resolved subclade. The incertae sedis genus Bulgariella also belongs in this family. The type species of the New Zealand endemic genus Chaetoscypha was recombined by Johnston (2002) as Pirottaea nidulans. Baral (2016) noted that morphologically P. nidulans has an ascus apical ring typical of Helotiaceae rather than Ploettnerulaceae, the family he accepted for Pirottaea. Sequences from a morphologically similar fungus from New Zealand, Pirottaea palmicola, support the Baral (2016) interpretation of these New Zealand “Pirottaea” species as Helotiaceae. Lachnaceae, the second family in the “helotioid clade”, forms a well resolved sister clade to the Helotiaceae.
The Han Clade 9/Stamnaria lineage/Vandijckellaceae clade includes a set of taxa with no obvious shared morphological or ecological features. Included here are specimens in the genera Cistella and Urceolella that Han et al. (2014) placed in their Clade 9 (both Hyaloscyphaceae sensu Baral 2016), Stamnaria (Stamnaria lineage sensu Baral and Haelewaters 2015, Baral 2016), the ex-type specimen of Vandijckella (Vandijckellaceae, Crous et al. 2017), along with the recently described Polyphilus (Ashrafi et al. 2018) and the type species of incertae sedis genera Tetracladium, Rommelaarsia, and Roseodiscus (Fig. 4). Baral (2016) discussed challenges with regard to morphological identification of some of the specimens referred to these genera. A targeted morphological and multi-gene molecular phylogenetic study is needed to resolve the taxonomy of this clade.
The informal “pezizelloid clade” includes the Pezizellaceae sensu Baral (2016) (this family includes specimens Han et al. (2014) placed in their Clades 1, 2, and 3), the genera treated by Han et al. (2014) as Clade 4 (this includes three of the genera placed in Hyaloscyphaceae by Baral (2016)), and the recently described mycorrhizal genus Gamarada (Fig. 4). The “pezizelloid clade” also contains a specimen identified as the type of Crocicreas, C. gramineum, and species in the cleistothecial genera Amorphotheca (Amorphothecaceae) and Oidiodendron (Myxotrichaceae). Fungi in Myxotrichaceae share the mycorrhizal lifestyle of Gamarada.
The “sclerotinioid” clade contains four strongly supported clades, Sclerotiniaceae + Rutstroemiaceae, Cenangiaceae, Cordieritidaceae and Chlorociboriaceae (Figs. 2 and 5). Although Sclerotiniaceae is well resolved as monophyletic, it makes Rutstroemiaceae as currently accepted paraphyletic. Based on the specimens we sampled, to retain monophyly within the Sclerotiniaceae + Rutstroemiaceae clade while retaining Sclerotiniaceae sensu Holst-Jensen et al. (1997), would require Rutstroemiaceae to be split into four separate families. Calycellinopsis, placed in Dermateaceae by Zhuang (1990), later in Helotiaceae by Zhuang et al. (2010) and then as incertae sedis in Baral (2016), belongs in Cenangiaceae. Also in the “sclerotinioid clade”, but not clearly within any of the families within the clade, are the genera Cryptohymenium and Polydesmia.
Leotiales as accepted here (Figs. 2 and 6) has a different concept to that of Baral (2016). It includes Leotiaceae, the newly described Mniaeciaceae, Tympanidaceae s. str., along with Claussenomyces (placed by Baral (2016) in a broader concept of Tympanidaceae), the recently described Aotearoamyces (Quijada et al. 2018), the aquatic hyphomycete genera Alatospora and Flagellospora, and another fungus that had been incorrectly placed in Collophorina. Mniaeciaceae is formally named here as a family to represent the Mniaecia lineage of Baral (2016, see below). Based only on the genera we sampled, Tympanidaceae is restricted to Tympanis, Collophorina, and Myriodiscus. The only other genera included in the family by Baral (2016) that we treated were Holwaya (here sister to Pseudeurotiaceae + Thelebolaceae in Thelebolales) and Grovesiella, here probably in Godroniaceae based on the ITS tree (Additional file 6: Figure S2), but on a questionably long branch.
Thelebolales are placed in a separate clade in Fig. 2. An alternative treatment would be to consider Pseudeurotiaceae and Thelebolaceae as morphologically highly specialised members of a genetically broader Leotiales. More intensive taxon sampling of the putative Leotiales genera discussed in the previous paragraph will be needed to resolve a defensible taxonomy for Thelebolales.
Phacidiales is here restricted to the Phacidiaceae and Helicogoniaceae. Within these families our analyses support the concepts of Baral (2016).
Rhytismatales in our analyses includes three families, Rhytismataceae, Cudoniaceae, and Triblidiaceae. A molecular phylogenetic relationship between Rhytismataceae and Cudoniaceae was recognized by Wang et al. (2002), and Magnes (1997) recognised Triblidiaceae as a member of the Rhytismatales based on a morphological study. The phylogenetic position of Triblidiaceae reported here is based on genes extracted from a genome from a single specimen (CBS 651.97) identified as Pseudographis elatina. This identification needs to be confirmed with additional sequence data from other specimens. Cudoniaceae forms a well resolved clade within a more inclusive Rhytismataceae + Cudoniaceae clade, but makes the remainder of Rhytismataceae paraphyletic. The morphologically based genera within Rhytismataceae have long been known to be incongruent with phylogeny (e.g. Lantz et al. 2011). The family requires a complete genus-level taxonomic revision. This revision must include resolution of the relationship between the families Rhytismataceae and Cudoniaceae.
Genera with enclosed ascomata have evolved independently several times across Leotiomycetes, but based on their distinctive ascomatal morphology they have often been treated as taxonomically distinct, despite being closely related in molecular phylogenetic studies to taxa with the disc-shaped ascomata more typical of Leotiomycetes. For example, the genus Loramyces is nested within Mollisiaceae, but retained as a separate family Loramycetaceae by Baral (2016) because of its unusual morphology, having enclosed ascomata with a perithecoid macromorphology. Other examples include Erysiphaceae with ascospores formed in small, enclosed, globose chasmothecia. They are generally treated as a separate order Erysiphales, despite being genetically members of Helotiales (Figs. 1 and 3). Connersia and Pleuroascus, with enclosed, cleistothecial ascomata have been placed in Pseudeurotiaceae, however genetically they appear to be morphologically specialised members of Helotiaceae. Bicornispora, originally placed in Coryneliales, is another cleistothecial member of Leotiomycetes, being closely related to the type species of Lambertella (Rutstroemiaceae), L. corni-maris (Additional file 6: Figure S2, Galán et al. (2015)). There is also strong molecular support for a sister relationship between the Thelebolales clade (sets of species in Pseudeurotiaceae and Thelebolaceae that have both apothecioid reduced cleistothecial ascomata) and the apothecial genus Holwaya and its asexual morph Crinula.
ITS gene tree
The ITS gene tree (Additional file 6: Figure S2) includes many more taxa than the 15-gene tree. Although deeper relationships are poorly supported in the ITS tree, many of the family level clades that were well supported in the 15-gene tree (Fig. 2) are also resolved in the ITS tree. These clades can help inform the taxonomic relationship of some genera that are not otherwise treated. Genera that were not treated in the 15-gene phylogeny but that are placed in the ITS tree with some confidence in one of the families or clades used in Fig. 2, and that had previously been referred to a different family (Additional file 7: Table S6), are discussed below.
The families Loramycetaceae, Mollisiaceae, Vibrisseaceae form with Strossmayeria and Chlorosplenium a well resolved clade in the ITS tree. Genera within this clade that were not treated in the 15-gene phylogeny and not previously placed in Mollisiaceae include Barrenia and Cheirospora (both previously incertae sedis), and Fuscosclera (previously Dermateaceae).
Ploettnerulaceae forms a well-resolved clade in the ITS tree. In the 15-gene tree it was represented only by the important plant pathogens Cadophora, Rhexocercosporidium, and Rhycosporium. Based on the ITS tree other genera in this family include the aquatic Collembolispora and the grass parasite Mastigosporium (both previously incertae sedis).
Godroniaceae forms a well-resolved clade in the ITS tree and this clade includes Grovesiella, previously referred to Tympanidaceae.
Gelatinodiscaceae splits into two clades in the ITS tree, one of these also includes Clathrosporium and Helicodendron (previously incertae sedis), and the type specimen of Dimorphospora (previously Helotiaceae). Neocudoniella, placed in Gelatinodiscaceae by Baral (2016), is in the Bryoglossum lineage in the ITS tree.
The Discinella-Pezoloma lineage forms a well-resolved clade in the ITS tree and this clade includes several additional aquatic hyphomycete genera —Cladochasiella, Fontanospora, Margaritispora, and Tetrachaetum — as well as specimens identified as the type species of Pseudopezicula (tentatively placed in the Drepanopezizaceae by Baral 2016) and Naevala (previously Calloriaceae).
Hyaloscyphaceae sensu our 15-gene analysis splits into two clades in the ITS tree. Hyaloscypha (including Meliniomyces, Pseudaegerita and Rhizoscyphus) forms a clade distant to that containing Hyalopeziza and Olla. Also in the Hyalopeziza ITS clade is a species of Arbusculina, but no data is available for type material of this genus. Unguicularia, accepted in Hyaloscyphaceae by Baral (2016), has an unresolved relationship in the ITS tree and awaiting additional genetic data is here accepted as Helotiales incertae sedis.
In an unresolved position in the 15-gene tree is a paratype specimen identified as “Roseodiscus” formosus, genetically distinct from the type species of Roseodiscus (Fig. 3). Based on the ITS tree, this specimen appears to belong in a well-supported clade with members of the Bryoglossum lineage sensu Baral (2016) that includes Bryoclaviculus, Bryoglossum, Neocudoniella, and “Crocicreas” multicuspidatum. Sampling of additional genes for more of these specimens may help resolve the position of the Bryoglossum lineage in the context of the current 15-gene analysis.
Arachnopezizaceae forms a well-resolved clade in the ITS tree. Sister to this clade is another with the aero-aquatic hyphomycete Clathrosphaerina and the phialophora-like Psychrophila. Support for this relationship is also indicated by LSU sequences for both genera, but not by the EF1a and β-tubulin sequences available for Psychrophila (unpubl. data). Two species in this clade referred to Durella, D. macrospora and D. melanochlora, are phylogenetically distinct from the type species of Durella, D. connivens (Baral 2016; Additional file 6: Figure S2).
Helotiaceae sensu our 15-gene analysis is resolved in the ITS tree. The core Helotiaceae clade in the ITS tree also includes Brunaudia (previously Patellariaceae), Endoscypha (previously Hyaloscyphaceae, and based on our ITS analysis possibly synonymous with Hymenotorrendiella), and Mitrulinia (previously incertae sedis). The “Hymenoscyphus” repandus specimen used for a genome study is closely related to the type species of Amylocarpus. As in the 15-gene tree, “Hymenoscpyhus” varicosporoides forms a separate subclade, and in the ITS tree this subclade includes additional aquatic hyphomycete genera Spirosphaera and Halenospora, along with Graddonia and Cudoniella, apothecial fungi reportedly associated with wet habitats (Hustad and Miller 2011).
The Han Clade 9/Stamnaria lineage/Vandijckellaceae lineage is spread across several clades in the ITS tree. Other genera that also belong in one or other of these ITS clades are Belonioscyphella (previously incertae sedis), Leohumicola (previously incertae sedis) and Mycoarthris (previously Hyaloscyphaceae). Also belong here are specimens representing three of the genera placed in Calloriaceae by Baral (2016), Calloria urticae (type species), Duebenia compta (type species) and Laetinaevia carneoflavida.
Pezizellaceae sensu our 15-gene analysis splits into three clades in the ITS tree. Also in one or other of these clades are Austropezia (previously Arachnopezizaceae), Porodiplodia (in the newly described family Porodiplodiaceae, Crous et al. 2018), and the previously incertae sedis genera Curviclavula, Hyalodendriella, Xenopolyscytalum, and Zymochalara.
Han Clade 4 from the 15-gene tree (Fig. 5) is well resolved in the ITS tree, except for Venturiocistella, a genus with no clear relationship in the ITS analysis. Based on the ITS tree, also in Han Clade 4 are Soosiella and Leptodontidium (both previously incertae sedis, and both sampled from the type specimen), and possibly Catenulifera, although this genus has no data available from the type species.
Cordieritidaceae mostly cluster in a poorly resolved clade in the ITS tree, and this clade includes the incertae sedis genera Sabahriopsis and Macroskyttea.
The ITS tree supports the congeneric relationship of Xylogone and Scytalidium suggested by Johnston et al. (2014). Only Scytalidium is treated in the 15-gene analysis and it has a poorly resolved position near the base of Helotiales.
Phacidiaceae forms a well-resolved clade in the ITS tree, and based on this tree two taxonomically poorly understood genera Epicladonia and Fulvoflamma might also belong in this family.
Based on our ITS tree several other genera could also possibly belong in the Leotiales sensu Fig. 6, including Cochlearomyces (Cochlearomycetacae, Crous et al. 2017), Gorgomyces, Miniancora, Mycosymbioses, and Satchmopsis. Patinella is close to Holwaya in the ITS tree. However, deeper relationships in this part of the ITS tree are poorly resolved and additional genes, additional taxa, and careful consideration of morphology is needed to develop a stable classification for Leotiales.
Other families not mentioned above that were sampled only with ITS data include Cyttariaceae, Heterosphaeriaceae, Mitrulaceae, and the recently described Neocrinulaceae. Based on the limited molecular data available, all occupy isolated positions within Helotiales, and whether or not they can be placed in one of our informal clades within Helotiales, or whether they form further distinct clades, requires increased gene sampling to resolve.
TAXONOMY
Marthamycetales P.R. Johnst. & Baral, ord. nov.
MycBank No.: MB827852
Etymology: named after the type genus Marthamyces.
Diagnosis: Phylogenetically isolated within Leotiomycetes; differs from the micro-morphologically similar Rhytismatales by the apothecia being deeply immersed in host tissue and having granular crystalline material on the hymenial surface.
Type: Marthamyces Minter 2003.
Description: Apothecia erumpent from substrate, covering layer splitting into irregular lobes to expose the more or less flat hymenial surface, underside of the lobes and surface of the hymenium often associated with a granular exudate giving a white, yellowish or greenish tinge. Excipular tissue layers reduced, marginal lobes often with internal periphysoids. Paraphyses simple or often highly branched near the apex. Asci thin-walled or with thick apical dome, nonamyloid. Ascospores 1-several septate, ellipsoid to filiform, lacking a gel sheath. Asexual morph unknown.
Habitat: Saprobic on wood, bark, and leaves, desiccation-tolerant.
Notes: Marthamycetales is proposed here as a new order with the single family Marthamycetaceae. Our results confirmed the comment by Baral (2015, 2016) that this family is phylogenetically isolated within Leotiomycetes.
Mniaeciaceae Baral, fam. nov.
MycoBank No.: MB828888
Etymology: named after the type genus, Mniaecia.
Diagnosis: Phylogenetically sister to Leotiaceae, differs by the small, sessile discoid apothecia and in being parasitic on liverworts.
Type: Mniaecia Boud. 1885.
Description: Apothecia gymnohymenial, sessile, superficial, non-gelatinous, white or blue-green, with smooth, non-protruding margin, hairless; ectal excipuletextura prismatica-globulosa, without crystals. Paraphyses simple, without vacuolar bodies. Asci with ± conical, inamyloid, thick-walled apex. Ascospores hyaline, broadly ellipsoid, non-septate, without sheath, with high lipid content (multiguttulate). Asexual morph unknown.
Habitat: Parasitic on liverworts on soil, desiccation-intolerant.
Included genera: Mniaecia (syn. Epiglia).
Notes: This new family was discussed by Baral (2016) as the Mniaecia lineage. It has a sister relationship with Tympanidaceae and Leotiaceae, and with the latter it shares the characteristics of lipid-rich ascospores and growth on soil.
Drepanopezizaceae Baral fam. nov.
Synonym: Drepanopezizaceae Bat. & H. Maia, Saccardoa 1: 98 (1960); nom. inval. (Art. 38.1)
MycoBank No.: MB828889
Etymology: named after the type genus, Drepanopeziza.
Diagnosis: Phylogenetically sister to Ploettnerulaceae, differs by the apothecia often being associated with stromatic structures; asexual morphs acervular, conidiogenesis holoblastic, and conidia large.
Type: Drepanopeziza (Kleb.) Jaap 1914.
Description: Apothecia sessile, immersed to erumpent, rarely superficial, often on stromatic tissue, hymenium greyish to brownish, margin often protruding, with or without lobes. Ectal excipule textura angularis. Paraphyses usually without vacuolar bodies. Asci with obtuse to conical apex, with or without amyloid apical ring. Ascospores ellipsoid to fusoid or often ± broadly ovoid-clavate, 0–1(− 2)-septate, septum ± eccentrical, lipid content low to medium. Asexual morph acervular, subcuticular, holoblastic, percurrent; conidia 0–1-septate, variously shaped, rarely staurosporous.
Habitat: Parasitic on leaves of various dicotyledons, causing leaf-spot diseases, rarely on herbaceous stems (Spilopodia); desiccation-tolerant.
Included genera: Blumeriella (syns. Higginsia, Phloeosporella, and Microgloeum), Diplocarpon (syn. Marssonina), Drepanopeziza (syn. Gloeosporidiella), Felisbertia, Leptotrochila (syns. Ephelina, Fabraea, and Sporonema), Pseudopeziza, Spilopodia (syns. Holmiodiscus and Melanodiscus), Spilopodiella, and Thedgonia.
Notes: The family was invalidly described (without a diagnosis or description) by Batista and Maia (1960) and is validated here. Our phylogenetic analyses support the treatment by Baral (2016) of this group of plant pathogenic fungi as distinct from the phylogenetically related Ploettnerulaceae. Pseudopezicula is excluded from the family as molecular data indicate affiliation in the Discinella-Pezoloma lineage.
DISCUSSION
The genomic and 15-gene phylogenies presented here are the first multi-gene, class-wide analyses of Leotiomycetes. They provide a framework for taxon and specimen selection for the future, targeted studies that are needed to formally develop a complete, modern classification of Leotiomycetes that is congruent with molecular phylogenetic relationships. The development of such a classification will require additional genomes to be sequenced to provide sufficient data to allow deeper relationships to be accurately resolved. Our phylogenies will inform the selection of taxa that are needed to be sampled at this level to provide that extra level of resolution. In addition, the data on which our phylogenies are based provide a set of reliable, taxonomically annotated DNA sequences that can be used in the meantime for placing specimens in a taxonomically informal, phylogenetic context. Such phylogenetic placement may help in understanding ecosystem function of unnamed organisms detected.
We propose only a few taxonomic novelties in this paper, because our analyses have limitations in terms of both taxon sampling and depth of gene coverage. Consequently, our results on their own are often inadequate to propose definite changes in the current classification. The groups of labelled clades defined in the phylogenies are based solely on molecular phylogenetic relationships. Where they are inconsistent with existing classifications, the source of this inconsistency (e.g. problems with our data, or problems with the current selection of taxonomically informative morphological characters) needs to be investigated in detail.
Three data quality issues might impact the reliability of the phylogeny presented as Figs. 2, 3, 4, 5 and 6. Firstly, gene coverage across the alignment is patchy. Only 30 of the specimens treated have all 15-genes available (Additional file 5: Table S7). Missing data such as this has been shown to affect accuracy of ML trees (e.g. Xi et al. 2016). Where two specimens have no genes in common in the data matrix, a pair-wise distance measure cannot be made. In our data matrix only 0.13% of the pairwise distance comparisons between specimens are missing. Three of our specimens lacked a pair-wise distance measure to more than 25 other specimens in the dataset. In none of these cases (KL299, KL332, and ERTZ16200) was their position in the phylogeny unexpected. Conversely, those specimens on long branches, such as 604a, D2514, and DH267, generally had very little missing data, with three or fewer between-specimen distances missing. This ad hoc examination of our data suggests that missing genes had minimal impact on the topology of the phylogenetic tree presented.
A second data quality issue that is potentially more problematic is the number and presumed placement of missing taxa. Theory (Townsend and Lopez-Giraldez 2010), as well as earlier analyses in this study that were based a smaller set of taxa (unpubl. data), demonstrated that the position of specimens on long branches is particularly impacted by the addition of extra, closely related specimens. Therefore, the position of several taxa on long branches, such as the type species of Cairneyella, Crocicreas and Cryptohymenium, could potentially be revised with molecular phylogenetic data from additional species within these genera, or from species of closely related genera. Many genera are represented by only ITS data. The placement of these within the classification accepted here should be regarded as tentative. The ITS analysis was included because it treats many more genera than the more data-rich analyses. Many of genera seem to have clearly resolved relationships in the ITS phylogeny, and the results of this analysis help highlight some of the outstanding taxonomic issues requiring additional sequence data. In addition, the ITS section of the paper provides a collation of reliable ITS sequence data (downloadable from the Manaaki Whenua Datastore) that are available to use to place sequences from unidentified specimens or DNA extracts in an approximate phylogenetic structure.
The third issue relates to the taxonomic names attached to specimens. Only 22 of the taxa in the 15-gene analysis are represented by type specimens or ex-type cultures. A further 84 genera are represented by specimens identified as the type species of that genus. Whether or not those identifications are correct has not been assessed in this study. Several genera are represented only by non-type species, that is species that may or may not be congeneric with the type of that genus.
The increasing number of genomes available for this class is providing an increasingly powerful taxonomic resource. The cost of sequencing a genome is continuing to drop, and the informatics tools now available make the production of a draft assembly of suitable quality for phylogenetic analyses straightforward. Of our analyses, only the genome-scale phylogeny provided strong support for the deepest, backbone branches in the phylogeny. It is these deep relationships that require resolution to provide support for possible new taxa at the ordinal level within the class Leotiomycetes. Despite the increasing number of genomes for Leotiomycetes, not every family has been sampled, and toward the base of the phylogeny, important for the resolution of the deep backbone (Townsend and Lopez-Giraldez 2010), sampling depth is particularly poor. A genome-based project targeting authentically identified specimens that represent poorly sampled parts of the trees presented here, should provide the data necessary to produce a really strong, phylogenetically sound classification. Future analyses of gene function based on these genomes will provide new insights into how effectively phylogeny, and classification based on that phylogeny, are able to predict lifestyle of newly discovered species with an unknown ecology. Ideally, such a classification will help predict the behaviour, or properties of interest, of ecologically poorly understood taxa included in the various clades resolved.
The Helotiales clade as defined in our analyses (Figs. 1 and 2) includes about 80% of specimens that we treat in both the 15-gene and ITS analyses. This high level of gene and taxon sampling reflects the predominance of Helotiales in the formal taxonomy of Leotiomycetes (Quijada 2018). If orders such as Cyttariales, Erysiphales and Medeolariales are accepted, then Helotiales is not only molecular phylogenetically extremely broad but is also highly paraphyletic. This paraphyly means that as a taxon it provides little useful information about relationships. However, the genome-scale phylogeny (Fig. 1), although based on a limited number of taxa, suggests that strong molecular phylogenetic structure does exist within the Helotiales clade. With increased taxon sampling of genome-scale data, underlying structure could provide the basis for a new, phylogenetically informative order-level taxonomy for Helotiales s.lat.
The treatment of paraphyletic relationships, often recognized taxonomically for sets of species with highly divergent, specialised morphologies, cannot be resolved by more informative phylogenies. Some taxonomists and users of classifications are willing to accept paraphyly within a classification (e.g. Davydov et al. 2010), to avoid ballooning numbers of taxonomic names of limited practical use. For example, strict monophyly within the Sclerotiniaceae + Rutstroemiaceae clade in our phylogeny, would require Rutstroemiaceae to be split into four or more families, if Sclerotiniaceae was to be retained. Sclerotiniaceae, a clade of genetically similar, economically important nectrotrophic pathogens, is an extremely useful taxonomic concept for plant pathologists. Because of its value to users, there are strong reasons for Sclerotiniaceae to be retained. The disadvantage is that the genetic diversity of the species within Rutstroemiaceae is ‘hidden’ by the classification. This may in turn inhibit an understanding of the evolution of lifestyles amongst these fungi, an understanding that is of economic significance when the fungi include plant pathogens, or mutualists advantageous to plant growth. Applied studies investigating the evolution of fungal lifestyles requires an accurate understanding of phylogeny to allow selection of an informative set of species. Where this selection is being applied by mycologists who are naïve to the taxonomic concepts applied, classifications that include paraphyletic taxa have the potential to mislead rather than to inform.
The phylogenies presented here are intended as a step toward a phylogenetically informative classification of Leotiomycetes. As discussed above, the DNA sequence data needed to generate strongly resolved phylogenies is incomplete. A cost- and time-effective way of providing the data needed for robust phylogenies across the class as a whole, would be through the sequencing of additional, selected genomes. The data generated will be of greatest value when that selection considers both the specimens being sequenced along with the informativeness of the taxon for phylogenetic uncertainties to be resolved. Our phylogenies provide a resource for the selection of those highly informative taxa for existing genera and families. This informed taxon selection should be combined with deliberate specimen selection, the most accurately identified specimen being the type specimen, and the most valuable type specimens are those available from public, curated fungaria and culture collections. To ensure Leotiomycetes classifications represent the global diversity of these fungi, there is a further need to actively sample taxa from poorly explored parts of the world. The study of fungal taxonomy has historically been largely based in North temperate regions, thus the current formal taxonomy unavoidably has a strong bias toward explaining the better catalogued north temperate diversity, a diversity that excludes important tropical and southern lineages.
CONCLUSIONS
The genomic and 15-gene phylogenies presented here provide a framework for taxon and specimen selection for the future targeted studies that are needed to formally develop a complete, modern classification of Leotiomycetes that is congruent with molecular phylogenetic relationships. The development of such a classification will require additional genomes to be sequenced to provide sufficient data to allow deep relationships to be accurately resolved. A strong, phylogenetically sound classification, combined with analyses of gene function based on the genomes used to resolve the phylogeny, will provide new insights into how effectively phylogeny, and classification based on that phylogeny, are able to predict lifestyle. Such a classification may help predict the behaviour, or properties of interest, of newly discovered species with an unknown ecology or other ecologically poorly understood taxa, and of unnamed taxa detected in ecological studies using molecular environmental sampling.
Abbreviations
- 5.8S :
-
Ribosomal DNA 5.8S region
- AFTOL-1:
-
Assembling the Fungal Tree of Life project
- EF1a:
-
Elongation factor 1-alpha gene
- IMC11:
-
11th International Mycological Congress in Puerto Rico
- ITS:
-
Ribosomal DNA internal transcribed spacer region
- LSU:
-
Ribosomal DNA large subunit region
- MCM7:
-
Minichromosome maintenance complex component 7 gene
- ML:
-
Maximum likelihood phylogenetic analysis
- mtSSU:
-
Mitochondrial small subunit gene
- NCBI:
-
National Center for Biotechnology Information
- RPA1:
-
Replication protein A 70 KDa DNA-binding subunit gene
- RPA2:
-
Replication protein A 32 KDa subunit gene
- RPB1:
-
DNA-directed RNA polymerase II subunit 1 gene
- RPB2:
-
DNA-directed RNA polymerase II subunit 2 gene
- RPC2:
-
DNA-Directed RNA polymerase III 127.6 KDa polypeptide gene
- SF3B1:
-
Splicing factor 3B subunit 1 gene
- SSU:
-
Ribosomal DNA small subunit region
- TFB4:
-
General transcription and DNA repair factor IIH subunit gene
References
Aberer AJ, Kobert K, Stamatakis A (2014) ExaBayes: massively parallel Bayesian tree inference for the whole-genome era. Molecular Biology and Evolution 31:2553–2556
Ashrafi S, Knapp DG, Blaudez D, Chalot M, Maciá-Vicente JG, Zagyva I, Dababat AA, Maier W, Kovács GM (2018) Inhabiting plant roots, nematodes, and truffles —Polyphilus, a new helotialean genus with two globally distributed species. Mycologia 110:286–299.
Banchi E, Ametrano CG, Stanković D, Verardo P, Moretti O, Gabrielli F et al (2018) DNA metabarcoding uncovers fungal diversity of mixed airborne samples in Italy. PLoS One 13(3):e0194489
Baral H-O (1992) Vital versus herbarium taxonomy: morphological differences beween living and dead cells of ascomycetes, and their taxonomic implications. Mycotaxon 44:333–390
Baral H-O (2015) Nomenclatural novelties. Index Fungorum 225:1–3
Baral H-O (2016) Inoperculate discomycetes. pp 157–205. In: Jaklitsch W, Baral H-O, Lücking R, Lumbsch HT, Frey W (eds) Syllabus of plant families: A. Engler’s syllabus der Pflanzenfamilien part 1/2. Borntraeger, Stuttgart, p 322
Baral H-O, De Sloover JR, Huhtinen S, Laukka T, Stenroos S (2009) An emendation of the genus Hyaloscypha to include Fuscoscypha (Hyaloscyphaceae, Helotiales, Ascomycotina). Karstenia 49:1–17
Baral H-O, Haelewaters D (2015) Rommelaarsia flavovirens gen. et sp. nov. (Helotiales), a new discomycete on Equisetum with a peculiar asexual state. Ascomycete.org 7:321–330
Baral H-O, Haelewaters D, Pärtel K (2015) A new attempt to classify the families of the Helotiales. In: The second international workshop on ascomycete systematics. CBS symposium, 22–24 April, Amsterdam
Baschien C, Tsui CK-M, Gulis V, Szewzyk U, Marvanová L (2013) The molecular phylogeny of aquatic hyphomycetes with affinity to the Leotiomycetes. Fungal Biology 117:660–672
Batista AC, Maia HS (1960) Discomycetes alienigenas (1). Saccardoa 1:98–102
Berbee ML (2001) The phylogeny of plant and animal pathogens in the Ascomycota. Physiological and Molecular Plant Pathology 59:165–187
Blehert DS (2012) Fungal disease and the developing story of bat white-nose syndrome. PLoS Pathology 8(7):e1002779
Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W (2010) Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27:578–579
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120
Castresana J (2000) Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Molecular Biology and Evolution 17:540–552
Chen C, Verkley GJ, Sun G, Groenewald JZ, Crous PW (2016) Redefining common endophytes and plant pathogens in Neofabraea, Pezicula, and related genera. Fungal Biology 120:1291–1322
Chernomor O, von Haeseler A, Minh BQ (2016) Terrace aware data structure for phylogenomic inference from supermatrices. Systematic Biology 65:997–1008
Coil D, Jospin G, Darling AE (2015) A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 31:587–589
Crous PV, Hawksworth DL, Wingfield MJ (2015) Identifying and naming plant-pathogenic fungi: past, present and future. Annual Review of Phytopathology 53:247–267
Crous PW, Quaedvlieg W, Hansen K, Hawksworth DL, Groenewald JZ (2014) Phacidium and Ceuthospora (Phacidiaceae) are congeneric: taxonomic and nomenclatural implications. IMA Fungus 5:173–193
Crous PW, Wingfield MJ, Burgess TI, Carnegie AJ, Hardy GESJ et al (2017) Fungal planet description sheets: 625–715. Persoonia 39:270–467
Crous PW, Wingfield MJ, Burgess TI, Hardy GESJ, Gené J et al (2018) Fungal planet description sheets: 716–784. Persoonia 40:240–393
Damm U, Fourie PH, Crous PW (2010) Coniochaeta (Lecythophora), Collophora gen. Nov. and Phaeomoniella species associated with wood necroses of Prunus trees. Persoonia 24:60–80
Davydov EA, Peršoh D, Rambold G (2010) The systematic position of Lasallia caroliniana (Tuck.) Davydov, Peršoh & Rambold comb. nova and considerations on the generic concept of Lasallia (Umbilicariaceae, Ascomycota). Mycological Progress 9:261–266
Day MJ, Hall JC, Currah RS (2012) Phialide arrangement and character evolution in the helotialean anamorph genera Cadophora and Phialocephala. Mycologia 104:371–381
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research 32:1792–1797
Eriksson OE (2005) Outline of Ascomycota. Myconet 11:1–113
Eriksson OE, Winka K (1997) Supraordinal taxa of Ascomycota. Myconet 1:1–16
Fehrer J, Réblová M, Bambasová V, Vohník M (2019) The root-symbiotic Rhizoscyphus ericae aggregate and Hyaloscypha (Leotiomycetes) are congeneric: phylogenetic and experimental evidence. Studies in Mycology 92:195–225
Galán R, Checa J, Blanco MN, Platas G, Tena R et al (2015) Taxonomic position of the genus Bicornispora and the appearance of a new species Bicornispora seditiosa. Mycologia 107:793–807
Gams W (2000) Phialophora and some similar morphologically little-differentiated anamorphs of divergent taxa. Studies in Mycology 45:187–199
Gnavi G, Ercole E, Panno L, Vizzini A, Varese GC (2014) Dothideomycetes and Leotiomycetes sterile mycelia isolated from the Italian seagrass Posidonia oceanica based on rDNA data. Singerplus 3:508
Grünig CR, Queloz V, Sieber TN (2011) Structure of diversity in dark septate endophytes: from species to genes. In: Pirttilä AM, Frank AC (eds) Endophytes of forest trees: biology and applications, forestry sciences 80. Springer, Dordrecht
Gulis V, Baschien C, Marvanová L (2012) Two new Tricladium species from streams in Alaska. Mycologia 104:1510–1516
Gurevich A, Saveliev V, Vyahhi N, Tesler G (2013) QUAST: quality assessment tool for genome assemblies. Bioinformatics 29:1072–1075
Hambleton S, Sigler L (2005) Meliniomyces, a new anamorph genus for root-associated fungi with phylogenetic affinities to Rhizoscyphus ericae (≡ Hymenoscyphus ericae), Leotiomycetes. Studies in Mycology 53:1–27
Han JG, Hosoya T, Sung GH, Shin HD (2014) Phylogenetic reassessment of (Hyaloscyphaceae sensu lato Helotiales, Leotiomycetes) based on multigene analyses. Fungal Biology 118:150–167
Hawksworth D (2001) The magnitude of fungal diversity: the 1.5 million species estimate revisited. Mycological Research 105:1422–1432
Hawksworth DL, Hibbett DS, Kirk PM, Lücking R (2018) (F-005-006) proposals to permit DNA sequence data to be used as types of names of fungi. IMA Fungus 9:v–vi
Hazard C, Gosling P, Mitchell DT, Doohan FM, Bending G (2014) Diversity of fungi associated with hair roots of ericaceous plants is affected by land use. FEMS Microbiology Ecology 87:586–600
Henríquez M, Vergara K, Norambuena J, Beiza A, Maza F et al (2014) Diversity of cultivable fungi associated with Antarctic marine sponges and screening for their antimicrobial, antitumoral and antioxidant potential. World Journal of Microbiology and Biotechnology 30:65–76
Hernández-Restrepo M, Gené J, Castañeda-Ruiz RF, Mena-Portales J, Crous PW, Guarro J (2017) Phylogeny of saprobic microfungi from southern Europe. Studies in Mycology 86:53–97
Hibbett D, Abarenkov K, Kõljalg U, Öpik M, Chai B et al (2016) Sequence-based classification and identification of Fungi. Mycologia 108:1049–1068
Hoang DT, Chernomor O, von Haeseler A, Minh BQ, Vinh LS (2018) UFBoot2: improving the ultrafast bootstrap approximation. Molecular Biology and Evolution 35:518–522
Holst-Jensen A, Kohn LM, Schumacher T (1997) Nuclear rDNA phylogeny of the Sclerotiniaceae. Mycologia 89:885–899
Hongsanan S, Jeewon R, Purahong W, Xie N, Liu JK et al (2018) Can we use environmental DNA as holotypes? Fungal Diversity 92:1–30
Hosoya T, Han J-G, Sung G-H, Hirayama Y, Tanaka K et al (2011) Molecular phylogenetic assessment of the genus Hyphodiscus with description of Hyphodiscus hyaloscyphoides sp. nov. Mycological Progress 10:239–248
Hosoya T, Sasagawa R, Hosaka K, Gi-Ho S, Hirayama Y et al (2010) Molecular phylogenetic studies of Lachnum and its allies based on the Japanese material. Mycoscience 51:170–181
Hustad VP, Miller AN (2011) Phylogenetic placement of four genera within the Leotiomycetes (Ascomycota). North American Fungi 6:1–13
Iturriaga T, LoBuglio KF, Pfister DH (2017) Bulgariella pulla, a Leotiomycete of uncertain placement, with an uncommon type of ascus opening. Mycologia 109:900–911
Johnston PR (2002) Chaetoscypha Syd. reassessed. New Zealand Journal of Botany 40:697–699.
Johnston PR, Seifert KA, Stone JK, Rossman AY, Marvanová L (2014) Recommendations on generic names competing for use in Leotiomycetes (Ascomycota). IMA Fungus 5:91–120.
Kalyaanamoorthy S, Minh BQ, Wong TFK, von Haeseler A, Jermiin LS (2017) ModelFinder: fast model selection for accurate phylogenetic estimates. Nature Methods 14:587–589
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution 30:772–780
Kauserud H, Mathiesen C, Ohlson M (2008) High diversity of fungi associated with living parts of boreal forest bryophytes. Botany 86:1326–1333
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M et al (2012) Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649
Kirk PM, Cannon PF, Minter DW, Stalper JA (2008) Ainsworth and Bisby’s dictionary of the fungi, 10th edn. CAB International, United Kingdom, p 771
Koizumi T, Nara K (2017) Communities of putative ericoid mycorrhizal fungi isolated from alpine dwarf shrubs in Japan. Microbes and Environments 32:147–153
Kühdorf K, Münzenberger B, Begerow D, Gómez-Laurito J, Hüttl RF (2015) Leotia cf. lubrica forms arbutoid mycorrhiza with Comarostaphylis arbutoides (Ericaceae). Mycorrhiza 25:109–120
Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology 10:R25
Lantz H, Johnston PR, Park D, Minter DW (2011) Molecular phylogeny reveals a core clade of Rhytismatales. Mycologia 103:57–74
Li H, Durban R (2009) Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25:1754–1760
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J et al (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2078
Liu Y, Whelen S, Hall B (1999) Phylogenetic relationships among ascomycetes: evidence from an RNA polymerse II subunit. Molecular Biology and Evolution 16:1799–1808
LoBuglio KF, Pfister DH (2010) Placement of Medeolaria farlowii in the Leotiomycetes, and comments on sampling within the class. Mycological Progress 9:36–368
Lorch JM, Lindner DL, Gargas A, Muller LK, Minnis AM, Blehert DS (2013) A culture-based survey of fungi in soil from bat hibernacula in the eastern United States and its implications for detection of Geomyces destructans, the causal agent of bat white-nose syndrome. Mycologia 105:237–252
Lumbsch HT, Huhndorf SM (2010) Outline of Ascomycota – 2009. Myconet 14:1–40
Lydolph MC, Jacobsen J, Arctander P, Gilbert MT, Gilichinsky DA et al (2005) Beringian paleoecology inferred from permafrost-preserved fungal DNA. Applied and Environmental Microbiology 71:1012–1017
Lόpez-Giráldez F, Townsend JP (2011) PhyDesign: an online application for profiling phylogenetic informativeness. BMC Evolutionary Biology 11:152
Magnes M (1997) Weltmonographie der Triblidiaceae. Bibliotheca Mycologica 165:1–177
Malloch D, Sigler L, Hambleton S, Vanderwolf KJ, Gibas CFC, McAlpine DF (2016) Fungi associated with hibernating bats in New Brunswick caves: the genus Leuconeurospora. Botany 94:1171–1181
Mitchell JI, Zuccaro A (2006) Sequences, the environment and fungi. Mycologist 20:62–74
Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ (2015) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Molecular Biology and Evolution 32:268–274
O’Brien HE, Parrent JL, Jackson JA, Moncalvo JM, Vilgalys R (2005) Fungal community analysis by large-scale sequencing of environmental samples. Applied and Environmental Microbiology 71:5544–5550
Pärtel K, Baral H-O, Tamm H, Põldmaa K (2017) Evidence for the polyphyly of Encoelia and Encoelioideae with reconsideration of respective families in Leotiomycetes. Fungal Diversity 82:183–219
Peng Y, Leung HCM, Yiu SM, Chin FYL (2012) IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28:1420–1428
Quijada L (2018) SIGS4 taxonomy of the Leotiomycetes – IMC11. In: The way fordward – a formal Leotiomycetes working group? International Mycological Congress, July 16–21, San Juan, Puerto Rico
Quijada L, Johnston PR, Cooper JA, Pfister DH (2018) Overview of Phacidiales, including Aotearoamyces gen. nov. on Nothofagus. IMA Fungus 9:371–382
Raja H, Miller A, Shearer C (2008) Freshwater ascomycetes: Aquapoterium pinicola, a new genus and species of Helotiales (Leotiomycetes) from Florida. Mycologia 100:141–148
Rokas A, Krüger D, Carroll SB (2005) Evolution: animal evolution and the molecular signature of radiations compressed in time. Science 310:1933–1938
Sanoamuang N, Jitjak W, Rodtong S, Whalley AJS (2013) Gelatinomyces siamensis gen. sp. nov. (Ascomycota, Leotiomycetes, incertae sedis) on bamboo in Thailand. IMA Fungus 4:71–87
Schoch CL, Sung G-H, López-Giráldez F, Townsend JP, Miadlikowska J et al (2009) Phylogeny clarifies the origin and evolution of fundamental reproductive and ecological traits. Systematic Biology 58:224–239
Schwessinger B, McDonald M (2017) High quality DNA from fungi for long read sequencing e.g. PacBio, Nanopore MinION, version 4. https://doi.org/10.17504/protocols.io.k6qczdw protocols.io
Sieber TN (2007) Endophytic fungi in forest trees: are they mutualists? Fungal Biology Reviews 21:75–89
Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31:3210–3212
Sivichai S, Jones EBG, Hywel-Jones NL (2003) Lignicolous freshwater Ascomycota from Thailand: Hymenoscyphus varicosporioides and its Tricladium anamorph. Mycologia 95:340–346
Spatafora JW, Sung GH, Johnson D, Hesse C, O'Rourke B et al (2006) A five-gene phylogeny of Pezizomycotina. Mycologia 98:1018–1028
Stenroos S, Laukka T, Huhtinen S, Döbbeler P, Myllys et al (2010) Multiple origins of symbioses between ascomycetes and bryophytes suggested by a five-gene phylogeny. Cladistics 26:281–300
Stewart EJ (2012) Growing unculturable bacteria. Journal of Bacteriology 194:4151–4160
Talavera G, Castresana J (2007) Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Systematic Biology 56:564–577
Taylor JW, Berbee ML (2006) Dating divergences in the fungal tree of life: review and new analyses. Mycologia 98:838–849
Thomsen PF, Willerslev E (2015) Environmental DNA – an emerging tool in conservation for monitoring past and present biodiversity. Biological Conservation 183:4–18
Toju H, Yamamoto S, Sato H, Tanabe AS, Gilbert GS, Kadowaki K (2013) Community composition of root-associated fungi in a Quercus-dominated temperate forest: “codominance” of mycorrhizal and root-endophytic fungi. Ecology and Evolution 3:1281–1293
Townsend JP, Lopez-Giraldez F (2010) Optimal selection of gene and ingroup taxon sampling for resolving phylogenetic relationships. Systematic Biology 59:446–457
Truong C, Mujic AB, Healy R, Kuhar F, Furci G et al (2017) How to know the fungi: combining field inventories and DNA-barcoding to document fungal diversity. New Phytologist 214:913–919
Varma A, Oelmüller R (2007) Advanced techniques in soil microbiology (Vol. 11, Soil Biology). Springer Berlin Heidelberg, Berlin, p 427
Wang Z, Binder M, Hibbett DS (2002) A new species of Cudonia based on morphological and molecular data. Mycologia 94:641–650
Wang Z, Johnston PR, Takamatsu S, Spatafora JW, Hibbett DS (2007) Towards a phylogenetic classification of the Leotiomycetes based on rDNA data. Mycologia 98:1065–1075
Wang Z, Nilsson RH, Lopez-Giraldez F, Zhuang W-Y, Dai Y-C et al (2011) Tasting soil fungal diversity with earth tongues: phylogenetic test of SATé alignments for environmental ITS data. PLoS One 6(4):e19039
Waterhouse RM, Seppey M, Simão FA, Manni M, Ioannidis P et al (2017) BUSCO applications from quality assessments to gene prediction and phylogenomics. Molecular Biology and Evolution 35:543–548
Wijayawardene N, Hyde KD, Lumbsch HT, Liu JK, Maharachchikumbura SSN, Ekanayaka AH et al (2018) Outline of Ascomycota: 2017. Fungal Diversity 88:167–263
Wijayawardene N, Hyde KD, Rajeshkumar KC, Hawksworth DL, Madrid H et al (2017) Notes for genera Ascomycota. Fungal Diversity 86:1–594
Xi Z, Liu L, Davis CC (2016) The impact of missing data on species tree estimation. Molecular Biology and Evolution 33:838–860.
Zamora JC, Svensson M, Kirschner R, Olariaga I, Ryman S et al (2018) Consideration and consequences of allowing DNA sequence data as types of fungal taxa. IMA Fungus 9:167–175
Zhao Y-J, Hosoya T, Baral H-O, Hosaka K, Kakishima M (2012) Hymenoscyphus pseudoalbidus, the correct name for Lambertella albida reported from Japan. Mycotaxon 122:25–41
Zhuang WY (1990) Calycellinopsis xishuangbanna gen. Et sp. nov. (Dermateaceae), a petiole-inhabiting fungus from China. Mycotaxon 38:121–124
Zhuang WY, Luo J, Zhao P (2010) The fungal genus Calycellinopsis belongs in Helotiaceae not Dermateaceae. Phytotaxa 3:54–58
Zijlstra JD, Pieter VH, Baar J, Verkley G, Summerbell RC et al (2005) Diversity of symbiotic root endophytes of the Helotiales in ericaceous plants and the grass, Deschampsia flexuosa. Studies in Mycology 53:147–162
Adherence to national and international regulations
Not applicable.
Ethics approval
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
The datasets generated and analysed during the current study are available in the Manaaki Whenua – Landcare Research Datastore, https://doi.org/10.7931/T5YV-BE95
Competing interests
The authors declare that they have no competing interests.
Funding
L.G.was supported by a postdoctoral fellowship through the “Beatriu de Pinós” from the Departament d’Universitats, Recerca i Societat de la Informació, Generalitat de Catalunya (ref. 2006 BP-A 10081). Z.W. and J.P.T. were supported by supported by National Science Foundation grants MCB-0923797 and IOS-1457044. K.P. was supported by the Estonian Science Agency (project IUT20–30) and the European Regional Development Fund (Centre of Excellence EcolChange). L.Q. thanks the support of “Fundación Ramón Areces”. W.-Y.Z. was supported by the National Natural Science Foundation of China (no. 31570018). P.R.J. and D.P. were supported through the Manaaki Whenua – Landcare Research Characterising Land Biota Portfolio with funding from the Science and Innovation Group of the New Zealand Ministry of Business, Innovation and Employment.
Authors’ contributions
All authors provided data toward the analyses presented, reviewed and approved the manuscript submitted.
Acknowledgments
We are grateful to the many authors acknowledged in the paper for the provision of publicly available fungal genomes. In addition, Francis Martin, the Mycorrhizal Genomics Initiative and the 1000 Fungal Genomes consortium, allowed access to unpublished genome data produced by the US Department of Energy Joint Genome Institute in collaboration with the user community; Mary Wildermuth and Shauna Somerville, University of California Berkeley provided access to genomes sequenced through the JGI CSP #1657 Comparative Genomics of Powdery Mildews and Associated Host Plants project, using biological material provided by Katherine Scheibel and Shauna Somerville (University of California, Berkeley); and Joseph Spatafora through the Assembling the Fungal Tree of Life funded by National Science Foundation (NSF). Paul Kirk (Royal Botanic Gardens, Kew) is thanked for providing downloads of selected taxonomic data from the Index Fungorum database. The New Zealand eScience Infrastructure (NeSI) provided computer support for the genome-scale phylogenetic analyses.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1:
Table S1. Specimens sampled, their taxonomic name, the family in which the genera were placed by Baral (2016) unless otherwise indicated, the family accepted based on the analysis in this paper, the sequences available for those specimens, either as Sanger sequences (with Genbank accesion number, otherwise available from alignments in https:/doi.org/10.7931/T5YV-BE95) or as extracts from genomes despoited in JGI and NCBI (Extracts available from alignments in https://doi.org/10.7391/T5YV-BE95). Newly generated sequences and genomes are in bold. (PDF 872 kb)
Additional file 2:
Table S2. Sequences used in ITS phylogeny, Additional file 6: Figure S2. Accepted family based on ITS and 15-gene analyses in this paper. (PDF 761 kb)
Additional file 3:
Figure S1. PhyDesign analysis. The upper image depicts the ultrametric tree for the taxa sampled using the single-copy orthologue dataset of Rokas et al. (2005) as modified by Taylor and Berbee (2006). Calibration points targeted the divergence of major Pezizomycotina lineages, approximately 250–400 million years ago, based on the fungal, animal and plant calibrations of Taylor and Berbee (2006). The lower image displays phylogenetic informativeness profiles for gene fragments from the datasets of Rokas et al. (2005), Taylor and Berbee (2006) and the AFTOL-1 project (Schoch et al. 2009). AFTOL-1 gene fragments are labeled with an asterisk (*). The five lesser-known fragments predicted to provide the greatest phylogenetic nformativeness for the desired epoch are also labeled. (PDF 456 kb)
Additional file 4:
Table S3. The top 10 loci estimated for effectively resolving major nodes basal in the Leotiomycetes phylogeny based on the PhyDesign analysis (Additonal file 3: Figure S1). Those used in our analyses marked with *. (DOCX 13 kb)
Additional file 5:
Table S7. Depth of gene coverage across the specimens sampled. (DOCX 12 kb)
Additional file 6:
Figure S2. ML tree based on ITS sequences. Labels include Genbank accession number, voucher number, family name accepted by Baral (2016) in brackets (as “( - )” if not treated in that work), and family name accepted on the basis of the 15-gene (Figs. 2, 3, 4, 5 and 6) and ITS phylogenies. Bootstrap values > 50% are indicated. The phylogenetic tree is rooted with Sarea and Tiarosporella. The alignment used for this analysis and a nexus version of the tree can be downloaded from the Manaaki Whenua – Landcare Research datastore, see https://doi.org/10.7931/T5YV-BE95. (PDF 7796 kb)
Additional file 7:
Table S6. Summary of the family level relationships of the genera treated, comparing the taxonomy of Baral (2016) with the relationships suggested in our analyses (genera where higher taxon has changed are in bold). Where a genus was not treated by Baral (2016) the family level classification provided by Index Fungorum in provided. Also provided is the type status of the specimens used in our analyses, and where the suggested relationship is based on the ITS analysis only. (PDF 566 kb)
Additional file 8:
Table S4. Basic data on the draft genome assemblies of 10 Leotiomycetes specimens deposited as NCBI Bioproject PRJNA487672. Data compares results from two assemblers. (DOCX 19 kb)
Additional file 9:
Table S5. Number of genes detected using BUSCO for each of the genomes used in genome phylogeny. Completeness value based on the BUSCO Pezizomycotina database (contains 3156 reference genes). (DOCX 19 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Johnston, P.R., Quijada, L., Smith, C.A. et al. A multigene phylogeny toward a new phylogenetic classification of Leotiomycetes. IMA Fungus 10, 1 (2019). https://doi.org/10.1186/s43008-019-0002-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43008-019-0002-x