
Abstract
Background
Helminth parasitism is a world-wide problem in livestock industries, with major impacts on health, welfare and productivity. The role of the gut microbiota in host-helminth interactions in ruminants has been extensively examined and the present study added to this body of knowledge by assessing the effects of resistance and susceptibility to helminth infection in the gastro-intestinal tract (GIT). Australian Sheep Breeding Values (ASBVs) for faecal egg count (FEC) were used to select the 10 highly helminth-susceptible (High-FEC) and 10 highly helminth-resistant (Low-FEC) sheep. FEC status was confirmed during the experiment. Using samples from the faeces and the lumen of the rumen, abomasum, duodenum, jejunum, ileum, caecum, and colon, DNA was extracted and used for 16 rRNA gene amplicon sequencing.
Results
The most frequent genera identified along the GIT were Eubacterium, Oscillibacter, and Ruminococcus. Intersectoral-specialization zones were identified along the GIT, with the duodenum displaying major differences between the High-FEC and Low-FEC animals in values for alpha and beta diversity. After taking all samples into account and adjusting for GIT segment, the High-FEC and Low-FEC sheep differed significantly for four genera Butyrivibrio, Mycoplasma, Lachnoclostridium and Succiniclasticum. In the duodenum, the abundances of Aminipila, Lachnoclostridium and Mogibacterium differed significantly between the High-FEC and Low-FEC sheep. In the ileum, on the other hand, the genus Mycoplasma was significantly depleted in the Low-FEC group.
Conclusions
The gastro-intestinal microbial profile varies widely between helminth-resistant and helminth-susceptible sheep. Each GIT section appears to support a particular bacterial composition leading to inter-sectoral differences among the various microbial communities. The microbial populations were most rich and diverse in the duodenum of helminth-resistant sheep, comprising bacterial genera that generally ferment carbohydrates. This observation suggests that helminth-resistant sheep can reorganize the duodenal microbiome taxa which may restrict the development of parasites.
Similar content being viewed by others


Background
Gastrointestinal helminth infection has a major economic impact in sheep industries worldwide—for example, for Australia in 2015, Meat and Livestock Australia estimated an annual loss of AUD436 million [1]. In the sheep of Western Australia, the major problem is infection during winter and early spring, with Teladorsagia circumcincta, found mainly in the abomasum, and Trichostrongylus colubriformis in the small intestine, particularly in the duodenum [2, 3]. Clinical signs associated with high levels of T. circumcincta and T. colubriformis infection include loss of appetite, rapid weight loss, profuse watery diarrhoea and hypoproteinaemia [4]. Other helminth species are also found, such as Chabertia ovina, Oesophagostomum venulosum and Haemonchus contortus, but they are less prevalent in this environment [5].
To mitigate helminth infection in their flocks, farmers worldwide have often resorted to frequent and intensive use of anthelmintic drugs, with the unfortunate outcome being the development of helminth populations that are resistant to one or more classes of parasiticide [6,7,8]. These problems have elicited global efforts in genetic selection using faecal egg count (FEC) as the phenotypic trait [9,10,11], to produce sheep that are naturally resistant to helminth infection. One example is the ‘Rylington Merino’ flock that was established in 1998 and is currently the most worm-resistant Merino flock in Australia [12]. In addition, the level of resistance to helminths varies within and between sheep genotypes, reflecting genetic variation in the production of immunoglobulin A (IgA) that is specific to helminth antigens, leading to variation in helminth survivability [13, 14].
There has been an increased interest in how microbial communities in the gastro-intestinal tract (GIT) are affected by parasite infection, raising the possibility of new avenues for investigating the drivers of gut homeostasis. For example, it was recently reported that the severity of infection with H. contortus in sheep is related to the faecal microbiota, implying a supporting role for gut microbes in modulating host resistance to infection [15]. Moreover, such observations suggest that, in the host, the intricate relationship between the immune system and the gut microbiome might be involved in the response to helminth infection [16]. In summary, it might be possible to identify particular bacterial communities that can be used to mitigate or control parasitic infection [17]. We therefore tested whether the structure and composition of the microbial communities along the GIT are affected by variation in genetic resistance to helminths, and identified specific microbial communities that are associated with helminth resistance. We used sheep with low (n = 10; Low-FEC) and high (n = 10; High-FEC) breeding values (genetic potential) for FEC and studied the GIT microbial populations using 16S rRNA gene amplicon sequencing.
Results
Parasitological differences between High-FEC and Low-FEC groups
The data for breeding value (ASBV), FEC and worm burden for the two genotypes are shown in Additional file 1: Table S1. The average values for cumulative FEC were 1940 ± 1120 eggs/g in the High-FEC group and 410 ± 423 eggs/g in the Low-FEC group (p = 0.018). This fourfold difference was a reflected in the average values for ASBV (High-FEC 35 ± 14 versus Low-FEC − 66 ± 4) and average values for worm burden (High-FEC 36,844 ± 23,733 worms versus Low-FEC 10,875 ± 12,320 worms). These observations validate the considerable divergence to helminth infection between the experimental groups.
General analysis of microbial communities along the GIT
As shown in Additional file 2: Table S2, a total of 13,100,180 raw reads were sequenced from the 16S V3-V4 amplicons generated from the faecal material and the luminal samples from the seven GIT segments of the 20 sheep. By trimming, merging of overlapping paired-end reads, and filtering of sequences < 400 bp, the number of reads was reduced to 3,013,423 sequences, ranging from 4,751 to 62,456 sequences per sample, and with an average sequence length of 449 ± 4 bp. These sequences were converted into 328 OTUs at 97% sequence identity, revealing 14 phyla, 22 classes, 27 orders, 39 families and 59 genera after taxonomic classification.
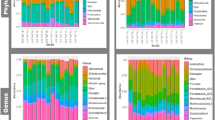
OTU richness and alpha diversity (Shannon index) of the microbial communities present in each GIT segment were calculated for High- and Low-FEC samples and statistical differences among the GIT segments of all sheep were assessed using ANOVA and Tukey’s post-hoc multiple comparisons analysis (Fig. 1 and Additional file 3: Table S3). The abomasum, rumen and duodenum contained the richest and most diverse populations, whereas the ileum produced the lowest values for both OTU richness and Shannon index (Fig. 1). Figure 2 shows significant differences in taxonomic composition between GIT segments (PERMANOVA, R2 = 0.564, p = 0.001). Principal coordinates analysis (PCoA) based on the weighted UniFrac distance presented a plot with a tight cluster containing the colon, caecum and faeces, with the rumen, abomasum, duodenum, jejunum and ileum in a different group with more scatter (Fig. 2). The segregation of the microbial communities was also evident in the contrast between the gastric (i.e., rumen and abomasum) and small intestine (i.e., duodenum, jejunum and ileum) compartments. Additional pairwise comparison revealed significant differences among all the GIT segments, except between caecum and colon (Additional file 6: Table S6). Firmicutes was the most abundant and ubiquitous phyla throughout the tract, followed by Bacteroidetes and Proteobacteria (Fig. 3). At genus level, Prevotella, Butyrivibrio, Saccharofermentans, Ruminococcus, Succiniclasticum, Desulfovibrio, Eubacterium, and Oscillibacter were the most common and ubiquitous (Fig. 4). The comparison of GIT segments shows that some phyla and genera are present or absent at specific sites and the most prominent differences are between the first part of the GIT (rumen to ileum) and the large intestine (caecum and colon). For example, Actinobacteria, Chloroflexi, Elusimicrobia, Euryarchaeota, Synergistetes, and Tenericutes are present exclusively in the first part of the GIT (Fig. 3). It should also be noted that eight genera (i.e. Treponema, Intestinimonas, Phascolarctobacterium, Anaeromassilibacillus, Anaerotignum, Paraprevotella, Flavonifractor, and Mailhella) were absent from the first part of the GIT, and 26 genera were absent from the large intestine (Fig. 4). The ANCOM test revealed 13 phyla and 52 genera that differed significantly among the GIT segments (Additional file 5: Table S5). Further statistical analysis, with each significant taxon compared among pairs of GIT segments is presented in Additional file 7: Table S7.

Differences in alpha diversity values, tested by ANOVA, among gastro-intestinal tract (GIT) segments from High- and Low-FEC sheep. a,b,cMeans followed by different letters above the graphs indicate statistically significant differences (p < 0.05). Graphs that share the same letter do not differ significantly

Principal coordinates analysis (PCoA) based on weighted UniFrac distance matrix of samples collected along the gastro-intestinal tract (GIT) of High- and Low-FEC sheep

Heatmap showing significant differences in bacterial abundance among gastro-intestinal tract (GIT) segments at phylum level, based on ANCOM analysis. Each value represents the median relative abundance in percentage

Heatmap showing significant difference in bacterial abundance among gastro-intestinal tract (GIT) segments at genus level, based on ANCOM analysis. Each value represents the median relative abundance in percentage
Analysis of the bacteria communities present in the High-FEC and Low-FEC groups
OTU richness and alpha diversity (Shannon index) were calculated for microbial communities present in each GIT segment, within and between FEC groups (Table 1). Interestingly, when the High-FEC and Low-FEC groups were compared, the only significant difference (p < 0.001) was the greater alpha diversity values for OTU richness and Shannon index in the duodenum of Low-FEC sheep compared to High-FEC. To avoid the potential confounding effect of FEC status, beta diversity across GIT segments was investigated separately for samples collected from High- and Low- FEC sheep. For the High-FEC group, our PCoA based on the weighted UniFrac distance metric (Fig. 5A) presented a plot with two tight clusters, one containing the rumen and abomasum samples, and the other containing the caecum, colon and faecal samples (PERMANOVA, R2 = 0.688, p = 0.001). Another group, comprising duodenum, ileum and jejunum, were more dispersed and well differentiated. For the Low-FEC group (Fig. 5B), there was a similar clustering effect (PERMANOVA, R2 = 0.601, p = 0.001), except that the duodenal samples had shifted towards the rumen-abomasum cluster, suggesting a different structure of duodenal microbiota in this group of animals. This observation prompted another round of PCoA to estimate differences in microbiota composition between the High-FEC and Low-FEC groups in the duodenum (Fig. 5C). The clustering differed significantly between the FEC groups (R2 = 0.583, p = 0.001). No differences were found for any of the other sites.

Principal coordinates analysis (PCoA) based on weighted UniFrac distance matrix data collected along the gastro-intestinal tract (GIT) of A the High-FEC group, B the Low-FEC group, C the High-FEC and Low-FEC groups in the duodenal segment
Table 2 presents the bacterial genera that differed significantly between the High-FEC and Low-FEC groups, for the total GIT and the individual GIT segments, as determined by ANCOM. In the duodenum, two genera, Aminipila and Lachnoclostridium, were significantly depleted, whereas Mogibacterium was enriched, in the High-FEC samples compared to the Low-FEC samples. In the ileum, on the other hand, there were significantly fewer mycoplasma in the Low-FEC group than in the High-FEC group. When taking all the samples into account, after adjustment for GIT segment, four bacterial genera differed significantly between the High-FEC and Low-FEC groups: the average relative abundances of Succiniclasticum and Butyrivibrio were significantly lower, whereas average relative abundances for Lachnoclostridium and Mycoplasma were significantly greater, in the High-FEC group compared with the Low-FEC group.
Discussion
This study compared GIT bacterial communities in helminth-resistant and helminth-susceptible sheep, in which there was a 400% difference in FEC and a 300% difference in parasite burden, following natural grazing under natural Mediterranean conditions [5]. The dominant helminths are Teladorsagia spp. in the abomasum and Trichostrongylus spp. in the duodenum. There were no significant differences between the groups of sheep for alpha diversity measurements in the rumen, abomasum, jejunum, ileum, caecum, colon or faeces. However, in the duodenum, OTU richness and Shannon diversity were lower in the susceptible sheep than in the resistant sheep. In general ecological terms, greater diversity in a community indicates a more stable and favorable environment. One possible explanation is that the duodenum of helminth-resistant sheep supports a better-adapted microbiome in which it may be difficult for helminths to thrive. Other factors that affect microbiome diversity within the gut include genetic background, physiological condition, diet, and health status [18]. In helminth-susceptible sheep, damage to the GIT by the parasite may inflame the duodenal mucosa, increasing protein leakage and changing the pH at this site, perhaps favoring the excessive growth and development of some bacterial species at the expense of others [19, 20]. The differences in alpha diversity observed across GIT segments were significant when both High- and Low-FEC groups were considered. Observed OTUs and Shannon index in the duodenum and jejunum (i.e., small intestine) were higher than in the large intestine. In this context, the lowest richness/diversity values were found in the ileum, suggesting that proliferation of the micro-flora is restricted in this segment of the small intestine, because of the high concentrations of bile, salts and digestive enzymes [21].
Three genera in the duodenum differed significantly in their abundance between the resistant and susceptible sheep—the Aminipila, Lachnoclostridium and Mogibacterium—the first two of which have only been recently identified as new taxa and were found abundant in the helminth-resistant group. In brief, Aminipila, isolated from cattle waste in 2018, has been associated with the degradation of L-arginine, L-lysine and L-serine, and with the production of short-chain fatty acids (SCFAs), particularly acetate and butyrate [22]. Lachnoclostridium, is a new genus that includes a number of new species that have been identified in the human gut in relation to colorectal tumorigenesis [23]. This genus was also significant in the helminth-resistant sheep when both High- and Low-FEC groups were examined along the whole GIT. They are associated with the production of butyrate [24, 25] but there is currently no information about their role or function in the sheep GIT. Recent evidence has suggested that specific fermenting bacteria are linked to intestinal homeostasis by the production of SCFAs metabolites associated to host metabolism, intestinal functions, and immunity system. For example, lower levels of butyrate were found in faeces of equines infected with high parasite burdens compared to the low parasite burden group [26]. Moreover, greater numbers of Mogibacterium were found in the duodenum of the helminth-susceptible group than in the helminth-resistant group, perhaps in association with disrupted gut conditions. This genus is abundant in humans with colorectal cancer, as well as in piglets fed a control formula that did not include beneficial prebiotics [27, 28]. Interestingly, the analysis of individual segments showed in the ileum of helminth-susceptible sheep a significant abundance of mycoplasma as well as along the whole GIT. Mycoplasmas are the smallest prokaryotic group found in nature and, as a result, they generally need host cells to supply biochemical compounds [29]. Many members of this genus have been identified in small ruminants, but only a few are considered to be clinically relevant [30]. At this point, the role of mycoplasmas in GIT responses to helminth infection is difficult to assess. Greater numbers of Succiniclasticum and Butyrivibrio differed in relative abundance in the helminth-resistant sheep along the whole GIT. This group mainly ferments carbohydrates to produce propionate, butyrate and formate. The genus Succiniclasticum, previously identified in the cow rumen, can only ferment succinate to produce propionate [31]. The members of the Butyrivibrio genus can produce, characteristically, butyrate and formate from a variety of carbohydrates [32]. In a previous study, Li and coauthors observed that helminth infection decreases the relative abundance of Butyrivibrio in goats infected with H. contortus [19]. These observations align with our finding of an increased abundance of butyrate-producing bacteria in helminth-resistant sheep, suggesting that SCFA-producing bacteria might be involved in the regulation of the helminth infection. Particularly butyrate, could regulate the helminth infection by reducing the expression of potent inflammatory molecules including tumor necrosis factor (TNF) and inducible nitric oxide synthase (iNOS) and regulate the recruitment and migration of immune cells (leukocyte, macrophages, dendritic cells, and T cells) to the infection site [33].
Microbial composition varied substantially among rumen-abomasum, small intestine, and large intestine, particularly with three genera: (i) Eubacterium, a genus that includes a wide spectrum of species [34]; (ii) Oscillibacter, an abundant genus in the faeces of free-grazing sheep (although with low values in the jejunum and ileum) that is linked to the production of butyric acid and alpha-linolenic acid [35]; and (iii) Ruminococcus, a genus of cellulose degraders found in various species [36]. We consider these three genera to be members of the core bacterial communities throughout the GIT in High- and Low-FEC sheep. These genera belong to the Firmicutes phylum and most of them can decompose fibre and cellulose [37]. Other studies in Small-Tailed Han and Chinese Mongolian sheep also describe Prevotella as one of the most important and ubiquitous genera throughout the sheep GIT [21, 38].
Conclusions
We evaluated and identified the microbiome from faeces and seven different sections along the sheep GIT and compared the populations in helminth-resistant and helminth-susceptible sheep. Every section of the sheep GIT supports quite specific bacterial communities and the distribution of communities is affected by resistance to parasite infection. This effect is particularly evident at the major sites of infection—specifically, the duodenum and the ileum. We conclude that helminth-resistant sheep support a more diverse variety of microbial communities and promote species that favor the production of SCFAs that could be involved in the processes that confer resistance to infection.
Methods
Animals and experimental design
This experiment was undertaken at the Katanning Research Station of the Department of Primary Industries and Regional Development (DPIRD) in Western Australia. It is located in a winter rainfall region with warm dry summers and cold wet winters. The most common and important worm species in this environment, T. circumcincta and T. colubriformis, can cause problems during winter, spring and up to early summer.
The sheep in this experiment were a sub-sample from a larger experiment [5] involving 986 lambs that had been born in July–August 2016 and weaned in November 2016. At weaning, the lambs were faecal sampled and administered a broad-spectrum oral anthelminthic (Monepantel; 1 mL/10 kg body weight). FEC was determined using the modified McMaster technique [39] with a sensitivity of 40 eggs/g faeces. Male and female lambs were separated at weaning and placed in two similar paddocks at a stocking rate of about 10 sheep/hectare. The pasture composition of the two fields during winter and spring was similar—primarily various Trifolium spp. and annual grass species with cape weed (Arctotheca calendula). In addition to the pasture, the sheep were supplemented with oaten hay ad libitum plus a supplement (500 g per sheep daily) of mixed barley and lupin grain, weaning until the end of the experiment in September 2017.
The FEC data at weaning, along with the completed pedigrees and the FEC data from previous generations, were submitted to Sheep Genetics [40], the Australian National Genetic Evaluation Scheme for sheep, to obtain the Australian Sheep Breeding Values (ASBVs) for FEC. ASBVs are estimated using Best Linear Unbiased Prediction mixed model methodology [41]. It uses all available pedigree information and adjusts the data for any factors that can affect the phenotypic measurement, such as management groups, animal sex, animal age, and birth status. The ASBV thus provides an unbiased prediction of the genetic worth of an animal, so it is the most accurate way to genetically differentiate between individuals and to identify genetically superior sheep. Additional information regards ASBVs can be found at following website operated by Meat and Livestock Australia [40].
Before the start of the experiment, the ASBVs for FEC were used to identify the most helminth-resistant and most helminth-susceptible animals: 100 males and 100 females (50 resistant and 50 susceptible sheep for each sex). These 200 sheep were maintained with their contemporaries for the duration of the experiment. From February 2017, they were faecal sampled monthly (Additional file 1: Table S1) to measure the increase in FEC up to slaughter in September 2017. In September 2017, we identified 18 sheep that had shown consistently high FEC and 20 sheep that had shown consistently low FEC, from February. These 38 sheep were transported to the laboratory in Albany in Western Australia. At 24 h prior to sacrificed, they were all confirmed to be in good health and were placed in individual sanitized pens with free access to water. After slaughter, the gut was immediately removed and luminal samples were collected from the rumen, abomasum, duodenum, jejunum, ileum, caecum, and colon. Faecal material was also sampled from each animal. All samples were stored at − 80 °C. From the 38 sheep, the 10 with the lowest FEC and 10 with the highest FEC were identified, and their luminal contents were analysed.
DNA extraction
DNA was extracted using the QIAamp® Fast DNA Stool Mini kit (Qiagen, Germany) with substantial modifications. In brief, 250 mg of each luminal or faecal sample was mixed individually with 1 mL of InhibitEX buffer and incubated at 95 °C for 5 min. The resultant supernatant was transferred to a new tube included 600 µL buffer AL and 25 µL of proteinase K incubated for 1 h at 70 °C. The cell lysate was thoroughly mixed with one volume of phenol:chloroform:isoamyl alcohol solution (25:24:1) for 1 min and centrifuged at 10,000 × g for 5 min. After recovering the aqueous phase, the process was repeated. Subsequently, the aqueous phase was transferred into a new 1.5 mL tube before adding an equal volume of chloroform:isoamyl alcohol solution (24:1). The mixture was vortexed for 1 min and centrifuged at 10,000×g for 5 min. The aqueous phase was again transferred into a new 1.5 mL tube, after which was added with two volumes of ice-cold 95% (v/v) ethanol to precipitate the DNA. The resulting DNA pellet was washed with 70% (v/v) ethanol and resuspended in 50 µL Tris–EDTA buffer (10 mM Tris–HCL, pH 8.0, 1 mM EDTA). DNA integrity was checked on a 1% (w/v) agarose gel electrophoresis and the amount of DNA was quantitated using a Nanodrop spectrophotometer.
16S rRNA gene library preparation
The V3-V4 hypervariable region of the 16S rRNA gene was amplified using the primer sets as specified in Illumina’s 16S metagenomic sequencing library preparation protocol [42]. In the initial round of PCR amplification, the reaction mixture contained 30 ng of input DNA, 2 units of Taq DNA polymerase (New England Biolabs, United States), 10 mM dNTP, 10 µM each of the forward and reverse primers, and 1 × standard Taq reaction buffer. The PCR conditions comprised an initial denaturation at 95 °C for 30 s, followed by 29 amplification cycles comprising denaturation (95 °C for 30 s), annealing (55 °C for 40 s), extension (68 °C for 1 min), and a final extension at 68 °C for 5 min. PCR amplicons were visualized using gel electrophoresis on a 1.5% (w/v) agarose gel. Following purification of PCR products using AMPure XP beads (Beckman Coulter, United States), indexing PCR was performed using Nextera® XT Index kit (Illumina, United States) according to manufacturer’s instructions. The libraries were sequenced on an Illumina MiSeq instrument using the 2 × 300 bp paired-end v3 chemistry.
Data analysis
Raw sequencing data were subjected to quality and adapter trimming using the bbduk.sh command available in BBTools (https://jgi.doe.gov/data-and-tools/bbtools/) with the following parameters: qtrim = r; trimq = 20; ktrim = r; k = 23; mink = 11; tpe; tbo; hdist = 1; and minlen = 200. After merging of overlapping paired-end reads using MeFiT software with default parameters, sequences with less than 400 bp were filtered [43]. The remaining sequences were subjected to de novo unoise clustering at 97% sequence identity threshold by running the micca otu command in Micca software (version 1.7.2) to obtain operational taxonomic unit (OTU) sequences [44]. Taxonomic classification of each representative OTU sequence was performed using the Bayesian LCA-based taxonomic classification method against the NCBI RefSeq 16S rRNA database [45], where the acceptance of a taxonomic assignment at each level was based on a minimum confidence score of 80. The OTU table and the taxonomic information are available in Additional file 4: Table S4.
Alpha and beta diversities were estimated using microbiomeSeq R package (https://github.com/umerijaz/microbiomeSeq) and QIIME v1.9.1 [46]. Following rarefaction at the sequence depth level of 1722, alpha diversity was analyzed on the basis of OTU richness and Shannon index metrics, and compared between groups using one-way analysis of variance (ANOVA) with Tukey's Honestly Significant Difference (HSD) post-hoc test. For analysis of beta diversity, principal coordinates analysis (PCoA) was performed at the OTU level using the weighted UniFrac measure and the statistical significance of the distance matrix was tested using permutational multivariate analysis of variance (PERMANOVA). Pairwise differences in beta diversity between GIT segments were calculated using beta-group-significance command implemented in QIIME2 v2021.11 [47]. For a variable to be considered having a significant influence on differences between groups in microbiota composition, a minimum R2 value of 0.25 and a p value less than 0.05 were both needed.
Differentially abundant taxa between groups
To identify bacterial phyla and genera that differed significantly among GIT segments, and between the high and low FEC groups within GIT segment, the analysis of composition of microbiomes (ANCOM) procedure [48] was performed on the raw abundance data using the ANCOM v2.1 R script (https://github.com/FrederickHuangLin/ANCOM). Bacterial taxa present in less than 15% of samples were excluded from the analysis. We adjusted the GIT segment comparisons for FEC level. The p values were adjusted using the Benjamini–Hochberg procedure at the significance level of 0.05. Bacterial taxa with significant associations were declared by using ANCOM’s W-statistic with a threshold of 0.7 (Additional file 5: Table S5). For each bacteria that differed significantly between GIT segments, further pairwise comparison were performed based on centered log-ratio (CLR)-transformed abundance data using Wilcoxon signed rank test with p values adjusted using the Benjamini–Hochberg method. The results are available in Additional file 7: Table S7.
Availability of data and materials
The raw sequencing reads generated in this study have been submitted to NCBI Sequence Read Archive (SRA) database under the BioProject accession number PRJNA674764.
Abbreviations
- ANCOM:
-
Analysis of composition of microbiomes
- ANOVA:
-
Analysis of variance
- ASBVs:
-
Australian sheep breeding values
- DPIRD:
-
Department of primary industries and regional development
- FEC:
-
Faecal egg count
- GIT:
-
Gastro-intestinal tract
- OTU:
-
Operational taxonomic unit
- PERMANOVA:
-
Permutational multivariate analysis of variance
- PCR:
-
Polymerase chain reaction
- PCoA:
-
Principal coordinates analysis
- SCFAs:
-
Short-chain fatty acids
References
Lane J, Jubb T, Shephard R, Webb-Ware J, Fordyce G. Priority list of endemic diseases for the red meat industries. Meat & Livestock Australia Limited; 2015. https://www.mla.com.au/contentassets/5c4a6eb332a94448b15602249c9fa6f1/b.ahe.0010_final_report.pdf. Accessed 30 Jul 2020.
Hutchinson GW. Nematode Parasites of Small Ruminants, Camelids and Cattle Diagnosis with Emphasis on Anthelmintic Efficacy and Resistance Testing Part 1. Diagnostic Overview. Aust New Zeal Stand Diagnostic Tech Proced Sub-Committee Anim Heal Lab Stand. 2009.
Makovcová K, Langrová I, Vadlejch J, Jankovská I, Lytvynets A, Borkovcová M. Linear distribution of nematodes in the gastrointestinal tract of tracer lambs. Parasitol Res. 2008;104:123–6. https://doi.org/10.1007/s00436-008-1169-z.
Taylor MA, Coop RL, Wall R. Veterinary parasitology. 4th ed. New Jersey: Wiley; 2015.
Greeff JC, Liu S, Palmer D, Martin GB, Greeff JC, Liu S, et al. Temporal changes in circulatory blood cell parameters of sheep genetically different for faecal worm egg count and diarrhoea from late summer to spring in a Mediterranean environment. Anim Prod Sci. 2020;60:1630–42. https://doi.org/10.1071/AN19038.
Ramos F, Marques CB, Reginato CZ, Bräunig P, Osmari V, Fernandes F, et al. Field and molecular evaluation of anthelmintic resistance of nematode populations from cattle and sheep naturally infected pastured on mixed grazing areas at Rio Grande do Sul. Brazil Acta Parasitol. 2020;65:118–27. https://doi.org/10.2478/s11686-019-00137-6.
Santiago-Figueroa I, Lara-Bueno A, González-Garduño R, López-Arellano ME, de la Rosa-Arana JL, Maldonado-Simán EJ. Anthelmintic resistance in hair sheep farms in a sub-humid tropical climate, in the Huasteca Potosin, Mexico. Vet Parasitol Reg Stud Rep. 2019;17: 100292. https://doi.org/10.1016/j.vprsr.2019.100292.
Lambertz C, Poulopoulou I, Wuthijaree K, Gauly M. Anthelmintic resistance in gastrointestinal nematodes in sheep raised under mountain farming conditions in Northern Italy. Vet Rec Open. 2019. https://doi.org/10.1136/vetreco-2018-000332.
Woolaston RR. Selection of Merino sheep for increased and decreased resistance to Haemonchus contortus: peri-parturient effects on faecal egg counts. Int J Parasitol. 1992;22:947–53. https://doi.org/10.1016/0020-7519(92)90052-M.
Bouix J, Krupinski J, Rzepecki R, Nowosad B, Skrzyzala I, Roborzynski M, et al. Genetic resistance to gastrointestinal nematode parasites in polish long-wool sheep. Int J Parasitol. 1998;28:1797–804. https://doi.org/10.1016/S0020-7519(98)00147-7.
Gauly M, Erhardt G. Genetic resistance to gastrointestinal nematode parasites in Rhön sheep following natural infection. Vet Parasitol. 2001;102:253–9. https://doi.org/10.1016/S0304-4017(01)00530-1.
Karlsson LJE, Greeff JC. Selection response in fecal worm egg counts in the Rylington Merino parasite resistant flock. Aust J Exp Agric. 2006;46:809–11. https://doi.org/10.1071/EA05367.
De Cisneros JPJ, Matthews L, Mair C, Stefan T, Stear MJ. The transfer of IgA from mucus to plasma and the implications for diagnosis and control of nematode infections. Parasitology. 2014;141:875–9. https://doi.org/10.1017/S0031182013002321.
Greer AW, McKenzie JL, McAnulty RW, Huntley JF, McNeilly TN. Immune development and performance characteristics of Romney sheep selected for either resistance or resilience to gastrointestinal nematodes. Vet Parasitol. 2018;250:60–7. https://doi.org/10.1016/j.vetpar.2017.12.013.
Mamun MA, Sandeman M, Rayment P, Brook-Carter P, Scholes E, Kasinadhuni N, et al. Variation in gut bacterial composition is associated with Haemonchus contortus parasite infection of sheep. Anim Microbiome. 2020;2:1–14. https://doi.org/10.1186/s42523-020-0021-3.
Mafuna T, Soma P, Tsotetsi-Khambule AM, Hefer CA, Muchadeyi FC, Thekisoe OMM, et al. Bacterial profiling of Haemonchus contortus gut microbiome infecting Dohne Merino sheep in South Africa. Sci Rep. 2021;11:1–11. https://doi.org/10.1038/s41598-021-85282-7.
Cortés A, Rooney J, Bartley D, Nisbet A, Cantacessi C. Helminths, hosts, and their microbiota: new avenues for managing gastrointestinal helminthiases in ruminants. Expert Rev Anti Infect Ther. 2020;18:977–85. https://doi.org/10.1080/14787210.2020.1782188.
Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220. https://doi.org/10.1038/NATURE11550.
Li RW, Li W, Sun J, Yu P, Baldwin RL, Urban JF. The effect of helminth infection on the microbial composition and structure of the caprine abomasal microbiome. Sci Rep. 2016;6:1–10. https://doi.org/10.1038/srep20606.
Besier RB, Kahn LP, Sargison ND, Van Wyk JA. The pathophysiology, ecology and epidemiology of haemonchus contortus infection in small ruminants. Adv Parasitol. 2016;93:95–143.
Wang J, Fan H, Han Y, Zhao J, Zhou Z. Characterization of the microbial communities along the gastrointestinal tract of sheep by 454 pyrosequencing analysis. Asian-Australasian J Anim Sci. 2017;30:100–10. https://doi.org/10.5713/ajas.16.0166.
Ueki A, Goto K, Kaku N, Ueki K. Aminipila butyrica gen. Nov., sp. Nov., a strictly anaerobic, arginine-decomposing bacterium isolated from a methanogenic reactor of cattle waste. Int J Syst Evol Microbiol. 2018;68:443–8. https://doi.org/10.1099/ijsem.0.002534.
Liang JQ, Li T, Nakatsu G, Chen YX, Yau TO, Chu E, et al. A novel faecal Lachnoclostridium marker for the non-invasive diagnosis of colorectal adenoma and cancer. Gut. 2020;69:1248–57. https://doi.org/10.1136/gutjnl-2019-318532.
Chen L, Zhou X, Wang Y, Wang D, Ke Y, Zeng X. Propionate and butyrate produced by gut microbiota after probiotic supplementation attenuate lung metastasis of melanoma cells in mice. Mol Nutr Food Res. 2021. https://doi.org/10.1002/MNFR.202100096.
Shuwen H, Miao D, Quan Q, Wei W, Zhongshan Z, Chun Z, et al. Protective effect of the “food-microorganism-SCFAs” axis on colorectal cancer: from basic research to practical application. J Cancer Res Clin Oncol. 2019;145:2169–97.
Peachey LE, Castro C, Molena RA, Jenkins TP, Griffin JL, Cantacessi C. Dysbiosis associated with acute helminth infections in herbivorous youngstock—observations and implications. Sci Rep. 2019;9:1–16. https://doi.org/10.1038/s41598-019-47204-6.
Berding K, Wang M, Monaco MH, Alexander LS, Mudd AT, Chichlowski M, et al. Prebiotics and bioactive milk fractions affect gut development, microbiota, and neurotransmitter expression in piglets. J Pediatr Gastroenterol Nutr. 2016;63:688–97. https://doi.org/10.1097/MPG.0000000000001200.
Chen W, Liu F, Ling Z, Tong X, Xiang C. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PLoS ONE. 2012;7: e39743. https://doi.org/10.1371/JOURNAL.PONE.0039743.
Razin S. The Genus Mycoplasma and Related Genera (Class Mollicutes). In: Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E, editors. The Prokaryotes: Volume 4: Bacteria: Firmicutes, Cyanobacteria. New York, NY: Springer US; 2006. p. 836–904. https://doi.org/10.1007/0-387-30744-3_29.
Nicholas RAJ. Improvements in the diagnosis and control of diseases of small ruminants caused by mycoplasmas. Small Rumin Res. 2002;45:145–9.
Van Gylswyk NO, Hippe H, Rainey FA. Pseudobutyrivibrio ruminis gen. nov., sp. nov., a butyrate-producing bacterium from the rumen that closely resembles Butyrivibrio fibrisolvens in phenotype. Int J Syst Bacteriol. 1996;46:559–63.
Palevich N, Kelly WJ, Leahy SC, Denman S, Altermann E, Rakonjac J, et al. Comparative genomics of rumen Butyrivibrio spp. uncovers a continuum of polysaccharide-degrading capabilities. Appl J Environ Microbiol. 2021. https://doi.org/10.1128/AEM.01993-19.
Yao Y, Cai X, Fei W, Ye Y, Zhao M, Zheng C. The role of short-chain fatty acids in immunity, inflammation and metabolism. Crit Rev Food Sci Nutr. 2022;62:1–12. https://doi.org/10.1080/10408398.2020.1854675.
Wade WG. The Genus Eubacterium and Related Genera. In: The Prokaryotes. Springer US; 2006. p. 823–35. https://doi.org/10.1007/0-387-30744-3_28.
Wang B, Luo Y, Su R, Yao D, Hou Y, Liu C, et al. Impact of feeding regimens on the composition of gut microbiota and metabolite profiles of plasma and feces from Mongolian sheep. J Microbiol. 2020;58:472–82. https://doi.org/10.1007/s12275-020-9501-0.
La Reau AJ, Suen G. The Ruminococci: key symbionts of the gut ecosystem. J Microbiol. 2018;56:199–208. https://doi.org/10.1007/S12275-018-8024-4.
Evans NJ, Brown JM, Murray RD, Getty B, Birtles RJ, Hart CA, et al. Characterization of novel bovine gastrointestinal tract Treponema isolates and comparison with bovine digital dermatitis Treponemes. Appl Environ Microbiol. 2011;77:138–47. https://doi.org/10.1128/AEM.00993-10.
Zeng Y, Zeng D, Ni X, Zhu H, Jian P, Zhou Y, et al. Microbial community compositions in the gastrointestinal tract of Chinese Mongolian sheep using Illumina MiSeq sequencing revealed high microbial diversity. AMB Express. 2017;7:1–10. https://doi.org/10.1186/s13568-017-0378-1.
Whitlock HV. Some modifications of the McMaster helminth egg-counting technique and apparatus. J Counc Sci Ind Res Aust. 1948;21:177–80.
Meat&LivestockAustralia. Sheep Genetics. https://www.sheepgenetics.org.au (2005). Accessed 25 Nov 2021.
Henderson C. Applications of linear models in animal breeding. Guelph: University of Guelph; 1984.
Illumina I. 16S metagenomic sequencing library preparation . Part # 15044223 Rev. B. https://support.illumina.com/downloads/16s_metagenomic_sequencing_library_preparation.html.
Parikh HI, Koparde VN, Bradley SP, Buck GA, Sheth NU. MeFiT: Merging and filtering tool for illumina paired-end reads for 16S rRNA amplicon sequencing. BMC Bioinformatics. 2016;17:491. https://doi.org/10.1186/s12859-016-1358-1.
Albanese D, Fontana P, De Filippo C, Cavalieri D, Donati C. MICCA: a complete and accurate software for taxonomic profiling of metagenomic data. Sci Rep. 2015;5:1–7. https://doi.org/10.1038/srep09743.
Gao X, Lin H, Revanna K, Dong Q. A Bayesian taxonomic classification method for 16S rRNA gene sequences with improved species-level accuracy. BMC Bioinformatics. 2017;18:1–10. https://doi.org/10.1186/s12859-017-1670-4.
Gregory Caporaso J, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6. https://doi.org/10.1038/NMETH.F.303.
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37:852–7. https://doi.org/10.1038/s41587-019-0209-9.
Mandal S, Van Treuren W, White RA, Eggesbø M, Knight R, Peddada SD. Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb Ecol Heal Dis. 2015. https://doi.org/10.3402/MEHD.V26.27663.
Acknowledgements
The authors thank Geoff Cox, Nicola Stanwyck and Tyler Loud who managed the research flocks at the Katanning Research Facility, and also the postgraduate students at the University of Western Australia who assisted with sampling the animals during this experiment.
Funding
EAP received postdoctoral fellowship support (reference number 74160038) from the National Commission for Scientific and Technological Research (Chile). The Department of Primary Industries and Regional Development of Western Australia is thanked for making funds available for this experiment.
Author information
Authors and Affiliations
Contributions
Conceptualization: GBM and CYT. Computational analysis and data interpretation: EGC and EAP. Funding acquisition: JCG and GBM. Investigation: EAP, JCG, SUH, DGP, SL, BL, NS, and JL. Writing (original draft): EAP and EGC. Writing (review and editing): GBM and JCG. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All animal experimentation was approved by the Animal Ethics Committee of the WA Department of Primary Industry and Rural Development, under the guidelines of the National Health and Medical Research Council’s Australian Code of Practice for the Care and Use of Animals for Scientific Purposes (approval no: AEC 17-1-02).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1
. Australian Sheep Breeding Values (ASBVs) for faecal egg count (FEC), worm counts and FEC of each animal used in the study.
Additional file 2: Table S2
. V3-V4 amplicons of the 16S rRNA gene generated from each sample included in the experiment.
Additional file 3: Table S3
. Results of Tukey’s test for post-hoc analysis of differences in microbial richness and Shannon index among gastro-intestinal tract (GIT) segments from High- and Low-FEC sheep.
Additional file 4: Table S4
. OTU table and taxonomic information. For each sample, relevant metadata [i.e., gastrointestinal segment and experimental group (High- or Low-FEC)] can be obtained from Table S2.
Additional file 5: Table S5
. ANCOM test used to the identification of bacterial phyla and genera showing significant differences among segments of the gastrointestinal tract, and between samples collected from High- and Low-FEC sheep.
Additional file 6: Table S6.
Pairwise differences in beta diversity among GIT segments of High- and Low-FEC sheep.
Additional file 7: Table S7.
Results of centered log-ratio (CLR) transformation using Wilcoxon signed rank test for each bacteria taxa that differed significantly among different GIT segments from High- and Low-FEC sheep.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Paz, E.A., Chua, E.G., Hassan, S.U. et al. Bacterial communities in the gastrointestinal tract segments of helminth-resistant and helminth-susceptible sheep. anim microbiome 4, 23 (2022). https://doi.org/10.1186/s42523-022-00172-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42523-022-00172-1