
Abstract
Background
The KCNT1 gene encodes a Na+-activated K+ channel. Gain-of-function mutations of KCNT1 lead to autosomal dominant sleep-related hypermotor epilepsy, early-onset epileptic encephalopathy, focal epilepsy and other epileptic encephalopathies. In this paper, we report a boy carrying a KCNT1 gene mutation, who presented with drug-resistant focal-onset seizures. He had decreased seizure frequency and improvement of background changes in electroencephalography (EEG) after vagus nerve stimulation (VNS).
Case presentation
The case was a nonverbal 9-year-old male who presented with drug-resistant focal-onset seizures since age 3 and had underwent VNS therapy for 2 years. He had hypermotor symptoms, automatism and bilateral asymmetric tonic seizures with cognitive decline and aphasis from age 3. The patient had a variety of seizure types that only occurred at night. The most common seizure type was automatisms, and ictal video EEG showed high-amplitude delta waves, followed by a fast rhythmic sharp activity in the mesial frontal and bitemporal regions. The patient was diagnosed with KCNT1-related epilepsy, epileptic encephalopathy and cognitive disorder. He was refractory to multiple anti-seizure medicines (ASM) and ketogenic diet. After VNS treatment at age 7, the frequency of seizures was reduced significantly and EEG was improved in background slowing.
Conclusions
Children with KCNT1-related epilepsy usually have early onset of disease, are nonverbal, and are refractory to ASM. This boy with drug-resistant KCNT1-related epilepsy showed significantly reduced seizure frequency after VNS. This report may provide reference for management of cases of KCNT1-related epilepsy.
Similar content being viewed by others

Background
KCNT1 is a gene localized at chromosome 9q34.3 in humans, which encodes a sodium-gated potassium channel. It is expressed diffusely in the brain, mainly in the cerebellum, frontal cortex and hippocampus, playing an important role in the regulation of neuronal excitability [1]. KCNT1 mutations, first described in 2012, have been found in epilepsy patients with different ages of onset and cognitive outcomes [2]. KCNT1 mutations are reported to cause developmental and epileptic encephalopathies (DEE), severe autosomal dominant sleep-related hypermotor epilepsy (ADSHE), focal temporal lobe epilepsy with intellectual disability and myoclonic-atonic epilepsy [1, 3,4,5]. Among the KCNT1-related DEE cases, half of them have malignant migrating focal seizures of infancy (MMFSI).
The KCNT1-related epilepsy seizures tend to be refractory to multiple anti-seizure medicines (ASMs) and ketogenic diet (KD), which show clinical efficacy in other epileptic encephalopathies [6,7,8]. Quinidine is expected to be an effective treatment for KCNT1-related epileptic encephalopathy by blocking the KCNT1 channel [9, 10]. However, recent studies have shown that quinidine treatment has no significant effectiveness [11]. Vagus nerve stimulation (VNS) is a nonpharmacologic therapeutic option for refractory epilepsy. VNS can improve cognition and reduce seizure frequency in patients with refractory epilepsy caused by genetic mutations, but its therapeutic mechanism remains to be determined [12].
Here, we describe a boy with refractory epilepsy caused by a KCNT1 mutation, who showed reduction in seizure frequency, as well as improvement of background changes in electroencephalography (EEG) and general condition after VNS.
Case presentation
This 9-year-old boy presented with drug-resistant focal onset seizures since age 3 and had multiple types of seizure semiology. Video EEG (VEEG) monitoring recorded 13 seizures, all of which occurred during stage 2 of sleep, including 1 hypermotor seizure, 9 automatism seizures and 3 bilateral asymmetric tonic seizures. The hypermotor seizures were manifested as trunck agitation and reptation movements, lasting less than 1 min. The ictal EEG showed onset from the bifrontal regions. The second seizure type was automatism seizures. The boy opened his eyes during sleep, with bilateral hand and mouth automatisms. The ictal EEG started with high-amplitude delta waves, followed by a fast rhythmic sharp activity on the left (in 8/9 attacks) and the right temporal lobes (1/9). The last seizure type was bilateral asymmetric tonic seizures characterised by head turning (1/3) and extension of the four extremities. This type of seizure had a frequency of about 3–4 times per night and was the main seizure semiology described by the parents. No aura was reported.
Interictal VEEG showed evidence of multifocal epileptic discharges including spike, multiple-spike and short-term fast rhythm bursts. Ictal VEEG suggested a multifocal onset. The patient was refractory to ASMs, including sodium valproate, levetiracetam and lacosamide, and allergic to oxcarbazepine.
The patient had no remarkable history of febrile convulsion, head injury or encephalitis. He suffered a severe degree of cognitive disorder and was nonverbal after the onset of epilepsy. He could only understand a few single words. The patient had no physical abnormalities during routine infant and childhood health examinations. Results of auxiliary examinations were also normal.
Genetic tests were conducted with consent from the patient and his family. The genetic testing revealed a novel mutation in the KCNT1 gene (chr9:138651532; c.862G > A; p.Gly288Ser) in this patient, while his parents were negative for mutation. This mutation was predicted to be pathogenic.
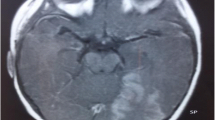
Magnetic resonance imaging (MRI) showed mild diffuse cerebral atrophy and focal cortical dysplasias (FCDs) in the right temporal lobe, and fluorodeoxyglucose positron emission tomography (PET) scan showed diffuse hypometabolism (Fig. 1).

Brain imaging: MRI scan revealed mild diffuse cerebral atrophy and focal cortical dysplasias in the right temporal lobe on T1-weighted (a) and FLAIR (b) images. PET-MRI showed hypometabolism in the right temporal-parietal area (c) and the bilateral frontal lobes (d)
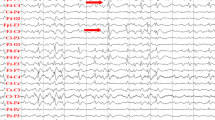
VNS was applied at age 7. The VNS settings were increased slowly, to reach a stimulation current output of 1.6 mA, a frequency of 30 Hz, a pulse width of 250 μs, a signal-on time of 30 s and a signal-off time of 5 min after 3 months. After 3 months of VNS using these parameters, the patient’s seizure frequency was markedly reduced from numerous seizures per day to several a week (Fig. 2). An improvement in background EEG was confirmed (Fig. 3). His parents reported an improvement in mood; however, there was no improvement in language function.

Monthly seizure activities. After VNS, the patient’s seizure frequency was reduced by 50% at the last follow up

Electroencephalographic monitoring. a At 7 years, immediately before VNS implantation, EEG showed central-occipital dominant background slowing. b At 8 years, ~ 1 year after VNS initiation, EEG showed improvement in background slowing
Discussion
The human KCNT1 gene, also known as Slack, was first molecularly described in 2000; it encodes a sodium-activated potassium channel [6]. KCNT1 is widely expressed throughout the brain, kidney and heart and is responsible for slow hyperpolarisation after action potential bursts. KCNT1 also interacts directly with fragile X-related proteins, and is involved in a highly extensive protein network, suggesting a putative role in cognitive-developmental processes [13].
KCNT1 gene mutations have been detected in various epileptic encephalopathies, such as West Syndrome, Lennox–Gastaut syndrome and MMFSI [14]. Sleep-related hypermotor epilepsy (SHE) with KCNT1 mutations was observed to have an earlier age of seizure onset and a severe intellectual disability. The mean age at seizure onset in KCNT1-related SHE was 60 months, while in non-KCNT1-related SHE it was more than 10 years. The KCNT1-related SHE usually has negative MRI findings and predominant hypermotor seizures [6]. Our case showed characteristic features of SHE: all of the seizures occurred during stage-2 sleep and the clinical expression consists of “hypermotor” events. However, the patient’s most common seizure type was automatisms, and MRI scan showed a lesion at the right temporal lobe. The temporal lobe epilepsy caused by KCNT1 mutations with a late onset has been reported recently [4]. The patient’s clinical features were partially compatible with both SHE and temporal lobe epilepsy. So he was more accurately diagnosed as KCNT1-related epileptic encephalopathies based on seizure symptoms and the clinical course [15]. Children with KCNT1-related epilepsy usually have an early onset, are nonverbal and refractory to ASM, and MRI findings include brain atrophy and FCD. A recent study showed that the p.Gly288Ser mutation can also cause SHE and MMFSI phenotypes [2], which means that there is no clear specific correlations between genotype and phenotype.
To date, at least 7 different genes have been shown to be associated with SHE, including KCNT1 [16]. Understanding the genetic aetiology could help us find new treatments [16]. Recent studies revealed that quinidine could block the KCNT1 channel and improve the electrophysiological abnormalities caused by KCNT1 mutations [17]. However, the reported efficacy of quinidine therapy has been contradictory [18]. A recent study showed that only 20% of patients have good response (> 50% reduction in seizures) [14]. The response to quinidine therapy may be age-dependent, as a study showed that only patients younger than 4 years have good response to quinidine treatment [19].
The MRI scan revealed mild diffuse cerebral atrophy and FCD at the right temporal lobe in the case. In KCNT1-related epilepsies, thin corpus callosum and brain atrophy are the most common findings from brain MRI. FCD induced by KCNT1 is rarely reported. FCD is commonly caused by gene mutations in components of the mTOR pathway. The exact mechanism of KCNT1-related FCD remains unclear to date. One study reported that patients with KCNT1-related focal refractory epilepsy have poor surgery outcomes despite having lesions (FCD I) on MRI [20]. KCNT1 mutations and FCD might reciprocally influence each other in the development of pathophysiology. In our patient, the MRI scan showed FCD at the right temporal lobe, but the PET scan showed more diffuse hypometabolism in the bilateral frontal lobes and the right temporal-parietal area. The PET abnormalities suggest that the gene is expressed diffusely in the central nervous system [21]. The functional imaging may help us assess the brain network in patients with KCNT1 mutations.
KCNT1-related epilepsy is often refractory to ASMs. KD is the most frequently reported treatment to reduce KCNT1-related seizures [6, 8], but it is difficult to implement in older children due to poor compliance. Sudden unexpected death in epilepsy (SUDEP) has been reported in patients with KCNT1-related ADSHE and MMFSI [7], which may be caused by the gene mutation in the heart.
VNS is a safe and effective neuromodulatory therapy for pediatric drug-resistant epilepsy, with a responder rate (> 50% seizure reduction) of around 35% to 50% after 2 years of follow up and higher rate after 5-year or longer follow-up [22, 23]. In children, the responder rate is higher than that in adults with drug-resistant epilepsy with any etiology [24]. The precise mechanism of VNS treatment remains unknown, although several hypotheses have been offered by previous studies [25]. Obviously, the mechanism is different from ASMs, which directly affect the ionic conductivity of neuronal membranes or affect the function of neurotransmitters.
Patients with genetic aetiology of drug-resistant epilepsy can also achieve significant outcomes after VNS. In tuberous sclerosis complex (TSC) patients, about 40% patients could acquire seizure freedom and the responder rate was 68% [24]. In contrast to TSC patients, the responder rate of Dravet syndrome with SCN1A mutation was only 41% and none were seizure free [12]. In Rett and Angelman syndrome, few cases respond to VNS [26].
The VNS therapy also improves the comorbidity of epilepsy such as cognitive and/or behavioral disorder, mental retardation, autism and attention deficit-hyperactivity disorder, independent of whether or not their seizures are controlled [27]. VNS may also reduce the rate of SUDEP. Ishii reported successful treatment of seizures with KCNT1-related MMFSI by VNS [28]. Our patient also responded to VNS, with 50% seizure reduction at the last follow up; however, whether it can benefit children with KCNT1-related epilepsy remains unknown.
Conclusions
Children with KCNT1-related epilepsy usually have an early onset, are nonverbal, and are refractory to ASMs. KCNT1-related focal refractory epilepsy has poor surgery outcomes. In this paper, we describe a boy with lesional drug-resistant KCNT1-related epilepsy, who showed significant improvement in seizure frequency after the initiation of VNS. Our report may provide reference for management of KCNT1-related epilepsy.
Availability of data and materials
Data are available upon reasonable request and are subject to restrictions imposed by patient confidentiality. All data are available upon request from the corresponding author.
Abbreviations
- ADSHE:
-
Autosomal dominant sleep-related hypermotor epilepsy
- EEG:
-
electroencephalography
- MMFSI:
-
Malignant migrating focal seizures of infancy
- MRI:
-
Magnetic resonance imaging
- VEEG:
-
Video electroencephalography
- VNS:
-
Vagus nerve stimulation
References
Møller RS, Heron SE, Larsen LH, Lim CX, Ricos MG, Bayly MA, et al. Mutations in KCNT1 cause a spectrum of focal epilepsies. Epilepsia. 2015;56(9):e114–20.
Lim CX, Ricos MG, Dibbens LM, Heron SE. KCNT1 mutations in seizure disorders: the phenotypic spectrum and functional effects. J Med Genet. 2016;53(4):217–25.
Barcia G, Fleming MR, Deligniere A, Gazula VR, Brown MR, Langouet M, et al. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat Genet. 2012;44(11):1255–9.
Hansen N, Widman G, Hattingen E, Elger CE, Kunz WS. Mesial temporal lobe epilepsy associated with KCNT1 mutation. Seizure. 2017;45:181–3.
Routier L, Verny F, Barcia G, Chemaly N, Desguerre I, Colleaux L, et al. Exome sequencing findings in 27 patients with myoclonic-atonic epilepsy: is there a major genetic factor? Clin Genet. 2019;96(3):254–60.
Bonardi CM, Heyne HO, Fiannacca M, Fitzgerald MP, Gardella E, Gunning B, et al. KCNT1-related epilepsies and epileptic encephalopathies: phenotypic and mutational spectrum. Brain. 2021;144(12):3635–50.
Kuchenbuch M, Barcia G, Chemaly N, Carme E, Roubertie A, Gibaud M, et al. KCNT1 epilepsy with migrating focal seizures shows a temporal sequence with poor outcome, high mortality and SUDEP. Brain. 2019;142(10):2996–3008.
Lin Z, Sang T, Yang Y, Wu Y, Dong Y, Ji T, et al. Efficacy of anti-seizure medications, quinidine, and ketogenic diet therapy for KCNT1-related epilepsy and genotype-efficacy correlation analysis. Front Neurol. 2022;12:834971 Published 2022 Jan 18.
Jia Y, Lin Y, Li J, Li M, Zhang Y, Hou Y, et al. Quinidine therapy for Lennox-Gastaut syndrome with KCNT1 mutation. A case report and literature review. Front Neurol. 2019;10:64 Published 2019 Feb 5.
Takase C, Shirai K, Matsumura Y, Watanabe T, Watanabe A, Hirasawa-Inoue A, et al. KCNT1-positive epilepsy of infancy with migrating focal seizures successfully treated with nonnarcotic antitussive drugs after treatment failure with quinidine: a case report. Brain and Development. 2020;42(8):607–11.
Numis AL, Nair U, Datta AN, Sands TT, Oldham MS, Patel A, et al. Lack of response to quinidine in KCNT1-related neonatal epilepsy. Epilepsia. 2018;59(10):1889–98.
Fulton SP, Van Poppel K, McGregor AL, Mudigoudar B, Wheless JW. Vagus nerve stimulation in intractable epilepsy associated with SCN1A gene abnormalities. J Child Neurol. 2017;32(5):494–8.
Cole BA, Clapcote SJ, Muench SP, Lippiat JD. Targeting KNa1.1 channels in KCNT1-associated epilepsy. Trends Pharmacol Sci. 2021;42(8):700–13.
Borlot F, Abushama A, Morrison-Levy N, Jain P, Puthenveettil Vinayan K, Abukhalid, et al. KCNT1-related epilepsy: an international multicenter cohort of 27 pediatric cases. Epilepsia. 2020;61(4):679–92.
Gertler T, Bearden D, Bhattacharjee A, Carvill G. KCNT1-Related Epilepsy. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews®. Seattle: University of Washington, Seattle; 2018.
Arenas-Cabrera C, Baena-Palomino P, Sánchez-García J, Oliver-Romero M, Chocrón-González Y, Caballero-Martínez M. Sleep-related hypermotor epilepsy with genetic diagnosis: description of a case series in a tertiary referral hospital. J Cent Nerv Syst Dis. 2022;14:11795735211060114 Published 2022 Feb 11.
Jia Y, Lin Y, Li J, Li M, Zhang Y, Hou Y, et al. Quinidine therapy for Lennox-Gastaut syndrome with KCNT1 mutation. A case report and literature review. Front Neurol. 2019;10:64 Published 2019 Feb 5.
Chong PF, Nakamura R, Saitsu H, Matsumoto N, Kira R. Ineffective quinidine therapy in early onset epileptic encephalopathy with KCNT1 mutation. Ann Neurol. 2016;79(3):502–3.
Abdelnour E, Gallentine W, McDonald M, Sachdev M, Jiang YH, Mikati MA. Does age affect response to quinidine in patients with KCNT1 mutations? Report of three new cases and review of the literature [published correction appears in Seizure. 2019 Jul;69:305]. Seizure. 2018;55:1–3.
Rubboli G, Plazzi G, Picard F, Nobili L, Hirsch E, Chelly J, et al. Mild malformations of cortical development in sleep-related hypermotor epilepsy due to KCNT1 mutations. Ann Clin Transl Neurol. 2018;6(2):386–91 Published 2018 Dec 25.
Fitzgerald MP, Fiannacca M, Smith DM, Gertler TS, Gunning B, Syrbe S, et al. Treatment responsiveness in KCNT1-related epilepsy. Neurotherapeutics. 2019;16(3):848–57.
Mao H, Chen Y, Ge Q, Ye L, Cheng H. Short- and long-term response of Vagus nerve stimulation therapy in drug-resistant epilepsy: a systematic review and Meta-analysis. Neuromodulation. 2022;25(3):327–42.
Pérez-Carbonell L, Faulkner H, Higgins S, Koutroumanidis M, Leschziner G. Vagus nerve stimulation for drug-resistant epilepsy. Pract Neurol. 2020;20(3):189–98.
Hajtovic S, LoPresti MA, Zhang L, Katlowitz KA, Kizek DJ, Lam S. The role of vagus nerve stimulation in genetic etiologies of drug-resistant epilepsy: a meta-analysis. J Neurosurg Pediatr. 2022;18:1–14.
Ohemeng KK, Parham K. Vagal nerve stimulation: indications, implantation, and outcomes. Otolaryngol Clin N Am. 2020;53(1):127–43.
Tomei KL, Mau CY, Ghali M, Pak J, Goldstein IM. Vagal nerve stimulation for medically refractory epilepsy in Angelman syndrome: a series of three cases. Childs Nerv Syst. 2018;34(3):395–400.
Toffa DH, Touma L, El Meskine T, Bouthillier A, Nguyen DK. Learnings from 30 years of reported efficacy and safety of vagus nerve stimulation (VNS) for epilepsy treatment: a critical review. Seizure. 2020;83:104–23.
Ishii A, Shioda M, Okumura A, Kidokoro H, Sakauchi M, Shimada S, et al. A recurrent KCNT1 mutation in two sporadic cases with malignant migrating partial seizures in infancy. Gene. 2013;531(2):467–71.
Acknowledgments
Not applicable.
Funding
This work was supported by Jinan Municipal Health Commission, 2019-2-29.
Author information
Authors and Affiliations
Contributions
MW, GG, HW and ZG analyzed and interpreted the patient data. YM and JS were major contributors in writing the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This research was approved by the Ethics Committee of Jinan Children’s Hospital and accorded with the Declaration of Helsinki (Register number: SDFE-IRBIT-2022034). Informed consent for clinical and genetic analyses was obtained from the parents prior to the study.
Consent for publication
The informed consent about publication was obtained from the patient’s parents.
Competing interests
All authors confirm that they have read the journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines. The authors have no personal, finance, or institutional interest in any of the drugs, materials, or devices described in this article.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, M., Geng, G., Meng, Y. et al. Long-term follow-up of vagus nerve stimulation in drug-resistant KCNT1-related epilepsy: a case presentation. Acta Epileptologica 4, 34 (2022). https://doi.org/10.1186/s42494-022-00105-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42494-022-00105-0