
Abstract
Background
Cerebral vasculopathies frequently lead to severe medical conditions such as stroke or intracranial hemorrhage and have a broad range of possible etiologies that require different therapeutic regimens. However, vasculopathies sometimes present with characteristic angiographic findings, that — if recognized — can guide a more specific diagnostic work-up. Certain ACTA2 variants are associated with a distinctive cerebrovascular phenotype characterized by an anomalously straight course of intracranial arteries, dilatation of proximal ICA and stenosis of distal ICA, in the absence of a compensatory basal collateral network found in Moyamoya disease. Until recently, this ACTA2 cerebral arteriopathy has been reported only in ACTA2 variants impairing Arg179.
Methods and materials
We report a familial case of a missense ACTA2 variant p.Arg198Cys with angiographic features of an ACTA2 cerebral arteriopathy. We analyzed the neuroimaging features of all four variant carrying family members and discussed the cerebrovascular abnormalities we found on the background of the current literature on ACTA2 arteriopathies.
Results
Neuroimaging of the variant carriers revealed angiographic abnormalities characteristic for ACTA2 cerebral arteriopathy such as stenoses of the terminal internal carotid artery, occlusion of the proximal middle cerebral artery and an anomalously straight course of the intracranial arteries. In our index patient catheter angiography showed a Moyamoya-like basal collateral network alongside with the above-mentioned features of an ACTA2 cerebral arteriopathy. The detected missense ACTA2 variant p.Arg198Cys was not known to be associated a cerebral arteriopathy, so far. One of the patients later died from aortic dissection — a common vascular complication of ACTA2 variants.
Conclusion
The familial case expands the phenotype of the detected ACTA2 variant p.Arg198Cys and hereby broadens the range of ACTA2 variants associated with a cerebral arteriopathy. Further, it emphasizes the importance of an interdisciplinary approach of vasculopathies.
Similar content being viewed by others


Background
Smooth muscle cell alpha-actin, encoded by ACTA2, is a key component of the contractile apparatus in smooth muscle cells of blood vessels and hollow organs [1]. Accordingly, heterozygous pathogenic variants of ACTA2 are associated with a wide spectrum of vascular but also systemic disorders. Thoracic aortic aneurysms and dissections (TAAD), occlusive vascular diseases such as juvenile strokes and coronary artery disease (CAD) are the most common and clinically relevant vascular manifestations of ACTA2 variants [2]. ACTA2 variants impairing Arg179 are associated with a particularly severe but also distinctive multisystemic phenotype summarized as syndrome of multisystemic smooth muscle dysfunction (SMDS) that exhibits congenital mydriasis, intestinal hypoperistalsis, hypotonic bladder, pulmonary hypertension, aortic disease and Moyamoya-like cerebral arteriopathy [2,3,4].
While in the past cerebrovascular abnormalities associated with ACTA2 variants were regarded as a variant of Moyamoya angiopathy (MMA), in recent years — due to its unique radiologic and histopathologic features — ACTA2 cerebral arteriopathy has been established as a novel entity independent of MMA [5]. Radiologic features of ACTA2 cerebral arteriopathy are a straight appearance of the intracranial arteries, dilatation of the proximal internal carotid artery (ICA) and stenosis of the distal ICA, in absence of the compensatory basal collateral network characteristic for Moyamoya disease [3,4,5]. Histopathologically, fibromuscular proliferation of the intimal and medial layer of cerebral arterial walls resulting in increased wall thickness and narrowing of the lumen might account for occlusive vascular complications as well as for the pathognomonic straight appearance of intracranial arteries in ACAT2 patients [1]. Fragmentation and loss of the elastic lamellae of the aorta might be causal for the increased risk of aortic aneurysms and dissection due to decreased resistance to blood pressure [1]. This distinctive cerebrovascular phenotype has first been described in patients with SMDS resulting from ACTA2 variants impairing Arg179 [4]. However, recently Kaw et al. published five cases of patients with ACTA2 variants and phenotypic features of SMDS, among whom four cases presenting variants not involving Arg179 [6]. This indicates that a broader range of ACTA2 variants manifest with features of SMDS. Consistent with this notion, features of ACTA2 cerebral arteriopathy have been observed in several variants that do not involve Arg179 (p.Arg258Cys, p.Arg258His, p.Asn117Lys, p.Asp181Val, p.Met46Arg) [6,7,8,9].
Methods and material
The clinical manifestations, medical history, laboratory and neuroimaging findings of four related carriers of the missense ACTA2 variant c.592C > T p.Arg198Cys were analyzed. Neuroimaging of all variant carrying family members was reviewed on the background of current literature on ACTA2 cerebral arteriopathy. For genetic testing DNA was isolated from whole blood samples of the patients. Based on the angiographic indication of an ACTA2 cerebral arteriopathy a targeted sequencing of the ACTA2 exons was performed.
Results/case description
In 2020, a 40-years-old woman was referred to our department of neurology for further diagnostic evaluation of the tentative diagnosis of MMA. The referral took place after the patient had developed recurring episodes of severe headache after physical activity and magnetic resonance imaging (MRI) had shown features of a unilateral variant of MMA with stenoses of the terminal right ICA, occlusion of the proximal right middle cerebral artery (MCA) and a basal collateral network, in the absence of white matter hyperintensities. Preexisting medical conditions were a malignant melanoma and a lumbar ependymoma — both successfully resected — as well as an umbilical hernia. Family history included amyotrophic lateral sclerosis in the patient’s father as well as the decease of her maternal grandmother at the age of 35 years due to unknown reason. At the time of proband’s assessment no family history of TAAD was recorded. Clinical examination revealed no neurologic deficits. Laboratory investigations including cerebrospinal fluid (CSF) analysis yielded no abnormalities of clinical relevance. The perfusion MRI revealed a cerebral perfusion deficit corresponding with the right sided MCA stenosis. In addition to the above-mentioned Moyamoya-like vascular features magnetic resonance and catheter angiography at our facility showed an anomalously straight course of the cerebral arteries suggestive of ACTA2 variants as well as a basal collateral network (Figs. 1a, b and 2a, b). Subsequently, genetic testing revealed a heterozygous ACTA2 variant c.592C > T p.Arg198Cys. The detected variant is listed in population databases with a minor allele frequency of 0.00001 (gnomAD). It has been submitted to ClinVar database four times in distinct subjects with TAAD. In the literature, this ACTA2 variant has previously been reported in two unrelated cases of early-onset TAAD, with occurrence of aortic events at 12 and 32 years of age [10]. In silico prediction tools (PolyPhen, SIFT) have indicated that the p.Arg198Cys variant disrupts protein function and classified it as probably deleterious. Based on its above-mentioned rarity in controls, recurrence in association with TAAD and in silico prediction of deleterious effects the p.Arg198Cys ACTA2 variant was classified as likely pathogenic in accordance with the American College of Medical Genetics and Genomics (ACMG) guidelines [11].

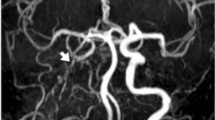
a Planar and b lateral view of the intracranial magnet resonance angiography (MRA) of the index patient showing Moyamoya-like features of the right middle cerebral artery (MCA) (arrow) as well as an anomalously straight course of the cerebral arteries; c planar and d lateral view of the intracranial MRA of the index patient’s mother also exhibiting straight cerebral arteries as well as bilateral stenosis of the terminal internal carotid artery (ICA) and proximal MCA (arrows); e planar and f lateral view of the intracranial MRA of the index patient’s daughter and son (g; h) with straight cerebral arteries

a Frontal and b lateral view of the catheter angiography of the index patient showing stenoses of the terminal right ICA and occlusion of the proximal right MCA (arrow with asterisk) as well as a Moyamoya-like basal collateral network (arrow with double asterisk); c MRI of the index patient’s mother showing white matter hyperintensities

Pedigree of the family. The index patient is indicated by an arrow. Empty symbols designate healthy individuals and full symbols designate family members affected by the p.(Arg198Cys) ACTA2 variant. Among the affected indivuals blackened symbols designate individuals with ACTA2 cerebral arteriopathy and blackened symbols with a white circle designate individuals with both ACTA2 cerebral arteriopathy and aortic dissection. Asterisks indicate individuals that underwent genetic screening
Due to the increased risk of aortic aneurysms and dissections associated with ACTA2 variants an MRI of the aorta was performed that ruled out aortic ectasia or aneurysm in our patient. Transesophageal echocardiography revealed the absence of a patent ductus arteriosus and ophthalmologic examination revealed absence of mydriasis as further abnormalities associated with ACTA2 variants. To minimize the risk of stroke indicated by impaired perfusion of the right MCA territory an extra- to intracranial bypass between the superficial temporal artery and MCA was operated and an antiplatelet therapy with 100 mg acetylsalicylic acid per day was initiated.
Genetic testing of the patient’s relatives revealed the same ACTA2 variant carried by the patient’s mother (61 years old; Fig. 3 II.2), her daughter (12 years old; Fig. 3 V.1) and one of her two sons (9 years old; Fig. 3 V.2). The fraternal twin brother of the affected son as well as the index patient’s maternal aunt were not found to be variant carriers. All mutated relatives were asymptomatic at the time of the genetic screening. The cranial MRI of the patient’s mother (Fig. 3 II.2) showed white matter hyperintensities (Fig. 2c) as well as features of ACTA2 cerebral arteriopathy such as the characteristic abnormally straight course of intracranial arteries, bilateral stenosis of the terminal ICA, proximal MCA and anterior cerebral artery (ACA) (Fig. 1c, d). Thoracic computed tomography (CT) revealed no aortic ectasia or aneurysm. However, the index patient’s mother suffered a dissection of the descending thoracic aorta in 2021 that was successfully treated by endovascular stenting. Later that year she died from aortic rupture. The cranial MRI of the index patient’s variant carrying children (Fig. 3 V.1., V.2) also showed the characteristic straight course of intracranial arteries (Fig. 1e–h). The children’s transthoracic echocardiography revealed the absence of a patent ductus arteriosus.
Discussion
Despite intensive diagnostic work-up the etiology of around 30% of strokes remains unknown [12]. Although rare, genetic disorders as ACTA2 variants are important to consider especially in juvenile and familial stroke cases which are not sufficiently explained by conventional risk factors. The described familial case expands the phenotype of the ACTA2 variant c.592C > T p.Arg198Cys, which formerly has been reported only in connection with TAAD in two individuals [10]. Our case is the first demonstrating the association of this missense variant with features of Moyamoya-like ACTA2 cerebral arteriopathy. ACTA2 cerebral arteriopathy is characterized by the pathognomonic straight course of intracranial arteries, dilatation of proximal ICA and stenosis of distal ICA, in the absence of a basal Moyamoya collateral network [3,4,5]. This distinctive cerebrovascular phenotype has been described first in patients with SMDS resulting from ACTA2 variants impairing Arg179 [4]. Recently, growing evidence has emerged suggesting that certain features of SMDS including ACTA2 cerebral arteriopathy do not exclusively occur in variants involving Arg179 but are present in a broader range of ACTA2 variants [6,7,8,9]. In the described case all four variant carrying family members exhibited features of ACTA2 cerebral arteriopathy. The particularly characteristic straight course of the intracranial arteries was present in all four variant carriers (Fig. 1a–h). The reported familial case hereby expands the genotype of ACTA2 cerebral arteriopathy. Notably, in our index patient alongside with characteristic features of an ACTA2 cerebral arteriopathy a Moyamoya-like basal collateral network was present, although not as highly developed as typically encountered in MMA (Figs. 1a, 2a, b) [13]. A dilatation of the proximal ICA — a repeatedly described feature of ACTA2 cerebral arteriopathy — was not present in any of the reported patients. This suggests a certain phenotypic heterogeneity of ACTA2 cerebral arteriopathy that might be attributed to the respective variant. However, an irregularly straight course of intracranial arteries is a highly recognizable radiologic feature that should prompt genetic testing for ACTA2 variants, even more so in patients who present several features of SMDS or a personal or family history of TAAD. However, the reported familial case demonstrates that genetic screening for ACTA2 variants should also be considered in Moyamoya-like arteriopathy with a straight course of intracranial arteries, even in the absence of a family history of TAAD or features of SMDS, and even if a basal collateral network is found. Identifying ACTA2 arteriopathy in patients suffering from neurological disorders and differentiating it from other cerebrovascular conditions such as cerebral atherosclerosis, vasculitis or MMA is of high relevance for the early detection of patients at risk of aortic dissection [5]. According to Milewicz and Regalado, early diagnosis of the variant and subsequent aortic surveillance, blood pressure control, and timely surgical repair of ascending aortic aneurysms, may help to reduce the risk of aortic complications [14, 15]. Notably, in ACTA2 variants, one third of the patients dissected at aortic diameters less than the diameter recommended for surgical repair of 5.0 cm. Therefore, surgical repair of aortic dilatations should be considered already at a diameter of 4.5 cm [14].
The reported case shows that the recognition of characteristic cerebrovascular abnormalities such as a straight course of intracranial arteries can guide the detection of an ACTA2 variant and this in turn can lead to the detection of a thoracic aortic aneurysm. Thus knowledge of the underlying genetic mutation can guide personalized aortic surveillance and hereby prevent aortic dissection and death. This case emphasizes the importance of an interdisciplinary approach of vasculopathies as the focus on only one organ system bears the danger of overlooking severe complications in others.
Conclusion
To the best of our knowledge this is the first time features of an ACTA2 cerebral arteriopathy have been found in patients carrying the p.Arg198Cys ACTA2 variant. Interestingly, a Moyamoya-like collateral network — usually absent in the context of ACTA2 variants — was found in our index patient, whereas a dilation of the proximal ICA — repeatedly described in other cases of ACTA2 cerebral arteriopathy — was not present in any of our patients. Although some variability can be observed in the cerebrovascular phenotype of distinct ACTA2 variants the anomalously straight course of the intracranial arteries is consistent and highly recognizable for ACTA2 cerebral arteriopathy. Another novelty of the reported familial case is the isolated nature of the Moyamoya-like arteriopathy in three out of four of our patients. Aside from the index patient’s mother who suffered an aortic dissection, no ACTA2 characteristics other than a Moyamoya-like arteriopathy were encountered in our patients, whereas ACTA2 mutated individuals with a Moyamoya-like arteriopathy reported in the literature typically present other ACTA2 features. Despite its rarity ACTA2 variants are important to diagnose as early detection can help to prevent severe vascular complications as aortic dissection, coronary artery occlusion and ischemic stroke.
Availability of data and materials
Not applicable as no datasets were generated or analysed.
Abbreviations
- ACTA2 :
-
Alpha actin 2
- ACA :
-
Anterior cerebral artery
- CAD :
-
Coronary artery occlusion
- CSF :
-
Cerebrospinal fluid
- CT :
-
Computed tomography
- ICA :
-
Internal carotid artery
- MCA :
-
Middle cerebral artery
- MMA :
-
Moyamoya angiopathy
- MRA :
-
Magnetic resonance imaging
- MRI :
-
Magnetic resonance angiography
- SMDS :
-
Multisystemic smooth muscle dysfunction
- TAAD :
-
Thoracic aortic aneurysms and dissections
References
Georgescu, M. M., Pinho, M. D. C., Richardson, T. E., et al. (2015). The defining pathology of the new clinical and histopathologic entity ACTA2-related cerebrovascular disease. Acta Neuropathologica Communications, 3(1), 81. https://doi.org/10.1186/s40478-015-0262-7
Guo, D. C., Papke, C. L., Tran-Fadulu, V., et al. (2009). Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and moyamoya disease, along with thoracic aortic disease. American Journal of Human Genetics, 84(5), 617–627. https://doi.org/10.1016/j.ajhg.2009.04.007
Milewicz, D. M., Østergaard, J. R., Ala-Kokko, L. M., et al. (2010). De novo ACTA2 mutation causes a novel syndrome of multisystemic smooth muscle dysfunction. American Journal of Medical Genetics Part A, 152A(10), 2437–2443. https://doi.org/10.1002/ajmg.a.33657
Munot, P., Saunders, D. E., Milewicz, D. M., et al. (2012). A novel distinctive cerebrovascular phenotype is associated with heterozygous Arg179 ACTA2 mutations. Brain, 135(8), 2506–2514. https://doi.org/10.1093/brain/aws172
Cuoco, J. A., Busch, C. M., Klein, B. J., et al. (2018). ACTA2 cerebral arteriopathy: Not just a puff of smoke. Cerebrovascular Diseases, 46(3–4), 159–169. https://doi.org/10.1159/000493863
Kaw, A., Kaw, K., Hostetler, E. M., et al. (2022). Expanding ACTA2 genotypes with corresponding phenotypes overlapping with smooth muscle dysfunction syndrome. American Journal of Medical Genetics Part A, 188(8), 2389–2396. https://doi.org/10.1002/ajmg.a.62775
D’Arco, F., Alves, C. A., Raybaud, C., et al. (2018). Expanding the distinctive neuroimaging phenotype of ACTA2 mutations. AJNR American Journal of Neuroradiology, 39(11), 2126–2131. https://doi.org/10.3174/ajnr.A5823
Mc Glacken-Byrne, A. B., Prentice, D., Roshandel, D., et al. (2020). High-resolution iris and retinal imaging in multisystemic smooth muscle dysfunction syndrome due to a novel Asn117Lys substitution in ACTA2: A case report. BMC Ophthalmology, 20(1), 68. https://doi.org/10.1186/s12886-020-01344-w
Pinto, M. (2020). A novel ACTA2 gene disease-causing variant presenting with a complex brain phenotype. Sinapse, 20(4), 181–183. https://doi.org/10.46531/sinapse/CC/200032/2020
Regalado, E. S., Guo, D., Prakash, S., et al. (2015). Aortic disease presentation and outcome associated with ACTA2 mutations. Circulation: Cardiovascular Genetics, 8(3), 457–464. https://doi.org/10.1161/CIRCGENETICS.114.000943
Richards, S., Aziz, N., Bale, S., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. https://doi.org/10.1038/gim.2015.30
Bersano, A., Kraemer, M., Burlina, A., et al. (2021). Heritable and non-heritable uncommon causes of stroke. Journal of Neurology, 268(8), 2780–2807. https://doi.org/10.1007/s00415-020-09836-x
Pilgram-Pastor, S., Chapot, R., & Kraemer, M. (2022). The angiographic presentation of European Moyamoya angiopathy. Journal of Neurology, 269(2), 997–1006. https://doi.org/10.1007/s00415-021-10684-6
Milewicz, D. M., & Regalado, E. S. (2015). Using genetics for personalized management of heritable thoracic aortic disease: how do we get there? The Journal of Thoracic and Cardiovascular Surgery, 149(20), S3–S5. https://doi.org/10.1016/j.jtcvs.2014.07.070
Milewicz, D. M., Regalado, E. S., & Guo, D. (2010). Treatment guidelines for thoracic aortic aneurysms and dissections based on the underlying causative gene. The Journal of Thoracic and cardiovascular Surgery, 140(6 Suppl), S2–S51. https://doi.org/10.1016/j.jtcvs.2010.07.027
Acknowledgements
Genetic analyses were carried out by Bioscientia GmbH, Ingelheim, Germany and Centogene GmbH, Rostock, Germany.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
JKF analysed and interpreted the clinical data, drafted and revised the manuscript. MK treated the patient, analysed and interpreted the clinical data, revised the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Given by the patients or their legal representatives.
Competing interests
JKF declares no conflict of interest. MK has received speaker honoraria from Roche Pharma and Chugai Pharma.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Focke, J.K., Kraemer, M. A familial missense ACTA2 variant p.Arg198Cys leading to Moyamoya-like arteriopathy with straight course of the intracranial arteries, aortic aneurysm and lethal aortic dissection. Neurol. Res. Pract. 5, 38 (2023). https://doi.org/10.1186/s42466-023-00268-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42466-023-00268-2