
Abstract
Sjogren’s disease (SjD) is an autoimmune disease that is characterized not only by the sicca symptoms it causes but also by its systemic nature, which is capable of several and not yet fully understood extraglandular manifestations. To gain a clearer understanding of these manifestations as well as a better practical approach, a panel of experts from the Brazilian Society of Rheumatology conducted a systematic review and meta-analysis on the identification of epidemiologic and clinical features of the extraglandular manifestations present in ESSDAI (EULAR Sjogren´s syndrome disease activity index), followed by a voting panel with recommendations for clinical practice. This publication is complementary to others already published and covers cutaneous and hematological manifestations, with prevalence data generated by a meta-analysis of 13 clinical or laboratory manifestations and 6 clinical management recommendations.
Similar content being viewed by others
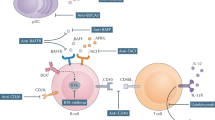


Background
Sjogren’s disease (SjD) is a systemic autoimmune disease, marked not only by the sicca symptoms it causes but also by its systemic nature, which is capable of various extraglandular manifestations [1, 2]. With an estimated worldwide prevalence of 0.01–0.05% [3, 4], its pathogenesis begins with glandular dysfunction resulting from the infiltration of T lymphocytes. This is followed by the activation of B lymphocytes and synthesis of autoantibodies capable of immune manifestations at a distance from the glandular structures [5].
Systemic extraglandular manifestations occur in around 40–50% of patients and can be severe. They include fatigue; myalgia; arthralgia and/or arthritis; gastrointestinal manifestations; and vascular, renal, pulmonary, neurological, cutaneous, or hematologic involvement. The severity of systemic involvement can be assessed using the ESSDAI (EULAR Sjögren’s syndrome disease activity index). In this tool, each manifestation mentioned corresponds to a domain. When systemic involvement is present, the patient receives a score that can vary from 0 to 2, 0 to 3, or 0 to 5 (depending on the domain). The sum of the affected domains generates the final ESSDAI value [6].
Despite being extensively validated and recommended for grading systemic involvement, the ESSDAI does not propose a routine diagnostic assessment for patients with SjD, which is an unmet need for physicians caring for patients with SjD. In addition, there are limited divergent data on the prevalence of systemic manifestations.
To address these gaps, the Sjögren’s Disease Committee of the Brazilian Society of Rheumatology conducted a broad systematic review of the literature on studies investigating extraglandular symptoms in Sjögren’s patients. The committee then gathered experts in the field and published recommendations for the screening and management of SjD patients with articular, pulmonary, renal, hepatic, gastrointestinal, and pancreatic manifestations [7, 8]. The current study represents an effort by the Brazilian Society of Rheumatology with the objective of retrieving the best available evidence and providing guidance for the identification of symptoms, diagnosis, monitoring, prognosis, and treatment of cutaneous and hematologic manifestations of SjD.
Methods
We conducted a systematic review of the diagnosis and prevalence of cutaneous and hematological manifestations in patients diagnosed with SjD according to the 2002, 2012, and 2016 classification criteria [9,10,11], following the recommendations proposed by the Cochrane Collaboration Handbook [12]. Questions were asked about the diagnosis and prevalence of cutaneous and hematological manifestations of SjD. An individualized search strategy for the different systemic manifestations was performed (Supplementary Material) for the Cochrane Central, MEDLINE, Embase, and LILACS databases.
The strategy was conducted with no restrictions on language or publication date. Only publications involving patients with the primary form of the disease (no other connective tissue disease) and those older than 18 years of age were selected. Observational studies in which the primary research question concerned the diagnosis and prevalence of individualized systemic manifestations were included. Articles that were duplicates across different databases were excluded along with those that did not concentrate on diagnosis and prevalence. To assess the diagnosis of systemic manifestations, preference was given to diagnostic accuracy studies. In the absence of such studies, we included any observational study that reported the use of diagnostic tests for detecting systemic manifestations of SjD.
The first study selection stage consisted of reading the titles and encompassing abstracts and keywords. The titles and abstracts of articles identified by the search strategy were independently assessed by two authors of the present study (AP and DCSE). In case of disagreement, a third author decided (VFMT). In the second stage, the investigators independently evaluated the full text of the articles and selected them according to pre-specified eligibility criteria.
To estimate the prevalence of cutaneous and hematological manifestations, a meta-analysis was carried out on the selected studies. We considered studies that specified both the number of patients affected by cutaneous or hematological manifestations and the total number of patients with SjD included. The statistical heterogeneity observed in these meta-analyses was anticipated by our review group, as it is a common occurrence in meta-analyses of prevalence. This expectation was due to the extensive range of patient characteristics, various available classification criteria (2002, 2012, and 2016), and different methods used to evaluate systemic manifestations. The risk of bias was evaluated using the Joanna Briggs Institute Prevalence Critical Appraisal Tool [13] (Supplementary Material). For the meta-analysis, we pooled clinical data by extracting the number of events and total number of patients to conduct a proportion meta-analysis. To estimate the overall proportion and present the pooled results with their respective 95% confidence intervals (CI), we employed a generalized linear mixed model (GLMM) method with a random-effects model. Calculations were performed using logit transformation in the “meta” and “metafor” packages from R software (version 3.6.1). Based on the data from the systematic review and meta-analysis, recommendations were formulated by Rheumatologists from the Sjögren´s Disease Committee of the Brazilian Society of Rheumatology. Agreement on these recommendations was achieved through online and in-person meetings using the Delphi Method [8].
Results
Figure 1 summarizes the steps involved in this systematic review. Important topics were described in sections reserved for cutaneous and hematologic manifestations, and based on these, two recommendations for cutaneous diseases and four recommendations for hematologic diseases were made (Table 1). Forest plots of the prevalence meta-analysis are shown at the end of each manifestation topic.

Flowchart of the studies selected for meta-analysis
Cutaneous manifestations
Skin involvement in SjD is relatively common and occurs in many patients. Several manifestations have been identified that significantly reduce patients’ quality of life by causing irritating symptoms, such as itching and pain. Table 2 summarizes the key points of the skin manifestations found in our systematic review.
The pathophysiological mechanism of mucocutaneous lesions in SjD is not fully understood, and an inflammatory response is usually identified [14, 15].
The classification of skin activity in ESSDAI [6] is based on the presence of certain types of skin lesions: absence of active skin lesions, 0 (no activity); erythema multiforme, 1 (low activity); limited cutaneous vasculitis, including urticarial vasculitis, purpura limited to feet and ankle, or subacute acute lupus erythematous (SCLE) = 2 (moderate activity); and diffuse cutaneous vasculitis, including urticarial vasculitis, diffuse purpura, or vasculitis-related ulcers = 3 (high activity).
The skin manifestation with the greatest clinical and prognostic importance associated with SjD is vasculitis, and patients may present with a wide variety of non-vasculitic lesions. Among the most common dermatological manifestations in SjD, we identified xeroderma, eyelid dermatitis, annular erythema, angular cheilitis, Raynaud’s phenomenon, and vasculitis, including nonpalpable purpura, livedo reticularis, urticaria, panniculitis, granuloma annulare, nodular localized cutaneous amyloidosis (NLCA), and lymphomatoid papules (LyP) [14, 16].
Xeroderma is a common condition characterized by dry scaly skin with reduced elasticity, usually causing itching and burning. This is the most frequent cutaneous manifestation of SjD, with studies showing its occurrence in 23–67% of cases [17,18,19]. The etiology of xeroderma is not fully understood. It was thought to be secondary to a reduction in sweat due to chronic inflammation of the exocrine glands; however, Bernacchi et al. did not report chronic inflammatory atrophy of sweat glands or reduced sweat rate, indicating that it was secondary to changes in the biochemical properties of the epidermis (increased epidermal proliferation and disrupted epidermal differentiation) [18, 20, 21]. Patients with xeroderma often develop rash complications with scratches on the skin and secondary hyperpigmentation [14, 17, 22]. In our meta-analysis, we identified a prevalence of 25% (95% CI 12–43%; 6 studies; 1144 participants) for xeroderma (Fig. 2).

Meta-analysis of xeroderma prevalence in Sjögren’s disease
Eyelid dermatitis is not a specific cutaneous manifestation of SjD and has been identified in approximately 40% of patients [23, 24]. It is often secondary to eye rubbing due to burning and itching caused by xerophthalmia [17, 24]. We identified a prevalence of 40% (95% CI 32–50%; two studies; 114 participants) of eyelid dermatitis (Fig. 3).

Meta-analysis of the prevalence of eyelid dermatitis in SjD patients
Annular erythema (AE) preferentially affects Asians and is rare in Caucasian patients. It is considered equivalent to SCLE lesions in whites [25, 26]. The first case of AE in a non-Asian population was described in 2014. It presents with lesions similar to erythema multiforme (erythematous lesions), often photosensitive, with a high border and clear center [27, 28]. Histopathology showed perivascular lymphocytic infiltration in 100% of the cases. AE occur more frequently in photo-exposed areas or in a disseminated form; however, the face is usually spared. It is strongly associated with positivity for anti-Ro/SSA and anti-La/SSB antibodies [27, 29]. AE is included in the cutaneous domain of the ESSDAI [6] and is classified as a manifestation of moderate activity [6, 30, 31]. In our Meta-analysis, we found a prevalence of 13% (95% CI 6–28%; 4 studies; 631 participants) (Fig. 4).

Meta-analysis of the prevalence of annular erythema in SjD patients
Angular cheilitis manifests itself as erythema with cracks and painful or painless sores in the corners of the mouth, secondary to anemia, or as a fungal or bacterial infection. It was identified as the second most frequent oral mucosal lesion in patients with SjD [32].
Raynaud’s phenomenon occurs in 20 to 35% of SjD patients, with a high incidence in those with cutaneous vasculitis [23].
Recommendation 1
-
Cutaneous vasculitis (palpable or nonpalpable purpura in the lower extremities and urticarial vasculitis) is associated with a higher risk of developing other extraglandular manifestations and lymphoma, especially in the presence of cryoglobulinemia and hypocomplementemia. In such cases, follow-up at smaller, regular intervals is recommended. (100% of agreement; Strength of Recommendation: Strong)
Recommendation 2
-
A biopsy can be performed for the differential diagnosis of skin manifestations. However, when the skin lesion is purpura, vasculitis can be clinically diagnosed. (93.4% of agreement; Strength of Recommendation: Strong)
Cutaneous vasculitis (CV) is the most important extraglandular manifestation in terms of prognosis and is associated with greater disease severity and systemic comorbidities [17]. It has been reported in up to 30% of SjD cases, and patients may present with a single episode or recurrent course [23, 33]. In our meta-analysis, we found a prevalence of 9% (95% CI 6–13%; 13 studies; 5659 participants) of this manifestation (Fig. 5).

Meta-analysis of the prevalence of cutaneous vasculitis in SjD
The most common clinical presentation is palpable or non-palpable lower-extremity purpura [33], followed by urticarial vasculitis. Less frequent lesions include erythematous macules, papules, ulcers, and ischemia [34]. CV predominantly affects small vessels, with rare reports of medium-vessel involvement. Histopathology shows leukocytoclastic vasculitis in 90% of cases; however, skin biopsy is not mandatory for the diagnosis of vasculitis associated with SjD [17, 23, 33,34,35,36]. Ramos-Casals et al found an association between low C4 levels and a high incidence of cutaneous vasculitis [37].
Waldenström’s hypergammaglobulinemia is a relatively common vasculitic manifestation in SjD, characterized by palpable purpura, marked polyclonal hypergammaglobulinemia, and high levels of rheumatoid factor and anti-Ro/anti-La antibodies [22, 33]. This condition is not usually associated with severe systemic disease but with peripheral neuropathy [38].
The presence of cryoglobulinemia with palpable purpura is associated with a higher prevalence of extraglandular disease and lymphoma, with high morbidity and mortality rates [37, 39, 40]. Cryoglobulinemia is associated with leukocytoclastic vasculitis in SjD in approximately 30% of the cases [34].
Laboratory markers of disease severity (low C3/C4 levels, positive rheumatoid factor, hypergammaglobulinemia, and cryoglobulins) were more frequent in patients with cutaneous vasculitis [34, 37].
The ESSDAI [6] classified the presence of vasculitis in SjD according to the extent of skin involvement (moderate activity when <18% and high activity when >18% of body surface area is involved) and the presence of ulcers (high activity).
There are reports of NLCA in SjD [41]. Other cutaneous lymphoproliferative disorders associated with SjD include LyP and cutaneous lymphomas. LyP is a benign condition in which 5–10% of cases progress to malignant lymphoma [42]. Other skin manifestations of SjD include vitiligo, alopecia, erosive lichen planus, erythema multiforme-like and erythema nodosum-like lesions, Sweet’s syndrome, granulomatous panniculitis, and subcorneal pustular dermatosis. However, these findings have been isolated and have unclear significance [22, 43, 44].
Hematological manifestations
For hematological manifestations, we will address the ESSDAI domain of the same name and the so-called biological domain (relating to laboratory alterations of the complement system, cryoglobulin positivity, and alterations in gamma globulins) since they intersect with hematology. Table 3 summarizes the key points of hematological manifestations found in our systematic review.
Recommendation 3
-
Blood counts should be assessed at least every 3–6 months (or earlier if necessary) in patients with SjD, since manifestations such as anemia, thrombocytopenia, and leukopenia (neutropenia and lymphopenia) are common. Before being attributed to disease activity, a broad evaluation of other causes should be performed. (100% agreement; Strength of Recommendation: Strong).
Leukopenia is one of the most important hematological manifestations [45, 46]. We have found a prevalence of leukopenia (<4000 cells/mm3), lymphopenia (<1500 cells/mm3), and neutropenia (<1500 cells/mm3) of 17% (95% CI 14 to 21%; 14 studies; 9074 participants), 12% (95% CI 5 to 24%; 6 studies; 2560 participants) and 12% (95% CI 5 to 23%; 5 studies; 2287 participants) respectively (Figs. 6, 7 and 8) [2, 45,46,47,48,49,50,51,52,53,54,55,56,57,58].

Meta-analysis of leukopenia in SjD

Meta-analysis of lymphopenia in SjD

Meta-analysis of neutropenia in SjD
The most likely mechanism is autoimmunity. As in other contexts, neutropenia is associated with greater severity given the risk of bacterial infectious complications.
Thrombocytopenia (<150,000 cells/mm3) was found in 3–14% of cases [46, 50], with an estimated prevalence of 8% (95% CI 6–12%; 11 studies; 7273 participants) in our meta-analysis (Fig. 9) [2, 45,46,47, 49, 50, 52, 53, 55, 57, 58]. A study evaluating the most frequent autoimmune etiologies of immune thrombocytopenia found SjD in 18.82% of cases, second only to systemic lupus erythematosus (SLE) [59].

Meta-analysis of thrombocytopenia in SjD
Anemia was the most prevalent manifestation, reaching 45% and with an estimated of 24% (95% CI 18 to 31%; 12 studies; 4963 participants in our meta-analysis (Fig. 10) [2, 45,46,47, 49, 50, 52, 53, 55, 56]. Zhou et al. reported that 69% of cases were classified as anemia of chronic disease, and 18% as autoimmune hemolytic anemia [53].

Meta-analysis of anemia in SjD
The ESSDAI presents a specific field in the hematological domain. In this field, only immune cytopenia were evaluated, and the score was given by the magnitude of the reduction in baseline values.
High activity is defined in the hematological field when hemoglobin, neutrophil, and platelet levels are below 8.0 g/dL, 500/mm3, and 50,000/mm3, respectively (one of these values is sufficient for the classification). There is no mention of lymphopenia in the criteria of high activity, as it presents a lower risk of complications than other cytopenias.
Recommendation 4
-
Complement C3 and C4 fractions and protein electrophoresis should be performed regularly as an additional step in assessing disease activity. (100% agreement; Strength of Recommendation: Strong).
The systematic review of the prevalence of complement consumption and serum cryoglobulin positivity also revealed clinically relevant data. When assessed in isolation, C3 consumption was present in 2–25% of cases [2, 60], and C4 consumption ranged from 5.0 to 37.2% [57, 60]. Our meta-analysis [2, 46,47,48,49,50, 52, 54,55,56,57,58, 60,61,62,63,64] found 12% (95% CI 9–17%; 16 studies; 15,467 participants) and 13% (95% CI 9–15%; 17 studies; 15,614 participants) of C3 and C4, respectively (Figs. 11 and 12).

Meta-analysis of the prevalence of C3 consumption in SjD

Meta-analysis of the prevalence of C4 consumption in SjD
Analysis from the Sjögren Big Data Project detected an association between complement consumption and a series of manifestations in other domains of ESSDAI (cutaneous, peripheral nervous system, and hematological) [63].
Regarding changes related to gamma globulins, the finding of hypergammaglobulinemia was frequent: 14 to 49% [57, 65, 66] with an estimated prevalence of 41% (95% CI 33 to 50%; 9 studies; 6311 participants) in our meta-analysis [2, 48, 50, 52, 54, 57, 64,65,66] (Fig. 13). In contrast, levels compatible with hypogammaglobulinemia showed much lower frequencies, varying from 2 to 6% [52, 66]. This study considered only disorders related to gamma globulin that were classified in a binary manner. Due to its diversity and low quantity, clonality characteristics were not evaluated in this systematic review.

Meta-analysis of hypergammaglobulinemia prevalence in SjD
The biological domain of ESSDAI includes gamma globulins and complements. There is no quantitative assessment of complement reduction; therefore, any decrease was classified as mild. Gammopathy disorders, however, can lead to the classification of mild or moderate activity.
Recommendation 5
-
Cryoglobulins should be measured at least once during follow-up (and regularly if the result is positive) and in the presence of manifestations suggestive of vasculitis (mainly peripheral neuropathy, glomerulonephritis, and purpura). (93.4% agreement; Strength of Recommendation: Conditional).
In the different studies, 3.6–28.0% of the patients were serum positive for cryoglobulin [54, 67] with an estimated prevalence of 12% (95% CI 9 to 15%; 17 studies; 10,034 participants) in our meta-analysis [2, 46,47,48, 52, 54, 56, 62, 63, 65, 67,68,69,70,71] (Fig. 14). The most common form of cryoglobulinemia in SjD is type 2 cryoglobulinemia. The positivity of cryoglobulinemia allows the classification of moderate activity in the biological domain of the ESSDAI, given the association of cryoglobulinemic vasculitis with higher overall mortality [56].

Meta-analysis of the prevalence of cryoglobulin in SjD
Recommendation 6
-
A systematized evaluation of lymphoma signs and symptoms must be performed in every follow-up consultation, especially in those presenting with risk factors. It should involve directed anamnesis (fever, night sweats, weight loss) and physical examination (palpation of the spleen, lymph nodes, and salivary glands). The presence of any suggestive findings in this evaluation merits prompt assessment for lymphomatous transformation, with those related to enlargement of the parotid glands or other salivary glands (chronic and/or asymmetrical enlargement and/or hardening/adherence and/or refractory to clinical measures) being of particular importance, as they are the most common site of lymphoma in patients with Sjögren’s. (100% agreement; Strength of Recommendation: Strong).
The risk of lymphoma in the population with SjD is higher than that in the population without the disease, or even in those with RA and SLE [72]. Because of the risk of lymphoma, the mortality associated with SjD is lower than that of the general population [67]. Classified as an ESSDAI manifestation, the possibility of lymphomagenesis in patients with SjD and its relationship with other hematologic manifestations deserves emphasis in these recommendations.
The main type of lymphoma found in SjD is the MALT (mucosa-associated lymphoid tissue) lymphoma of the salivary gland. This is a B-lymphocyte lymphoma, non-Hodgkin’s lymphoma, classified as an extra-nodal marginal zone (ENMZ) lymphoma. Diffuse B-Cell Lymphoma (DBCL) and B lymphoma of the Nodal Marginal Zone (NMZ) are the second and third most common types of lymphoma, respectively [73].
A few steps have been hypothesized to explain the lymphomagenesis that occurs in SjD. Upon stimulation by cytokines and other substances, an increase in the pool of B lymphocytes in the glandular epithelial unit occurs. Oncogenes and tumor suppressor genes drive polyclonal B-lymphocytes to undergo a shift to monoclonality, the hallmark of lymphoma [74]. The transformation of a primary MALT lymphoma into a DBCL or NMZ could explain the high prevalence of these types in SJD [75, 76].
It is possible that there is an effect of the duration of the disease, since it takes an average of 6.5 years for lymphoma to appear, and that there is a cumulative risk as high as 18% in those with 20 years of disease [77]. Persistent parotid enlargement, C4 consumption, cryoglobulinemia, rheumatoid factor, and disease activity are often considered risk factors for lymphoma in SjD [74] and reflect the importance of the salivary gland epithelium and B lymphocytes in lymphomagenesis.
Persistent and/or asymmetric enlargement of the salivary glands (especially the parotid gland) is the main clinical finding that needs an evaluation directed towards the possibility of lymphoma [78]. In such cases, imaging evaluation and discussion with specialists are recommended for the evaluation of incisional or excisional biopsy. Fine needle aspiration is not recommended [79,80,81].
Conclusion
In the third part of a series of systematic reviews and meta-analyses on the extraglandular manifestations of SjD conducted by the Brazilian Society of Rheumatology, prevalence data on cutaneous and hematological manifestations were generated, as well as six recommendations for the practical evaluation and diagnosis of these manifestations. The heterogeneity of the data found here points to the need for larger prospective epidemiological studies on the cutaneous and hematological manifestations of SjD, which will allow for a larger and more precise number of recommendations for future clinical practice.
Data availability
All data generated or analyzed during this study are included in this published article [and its Additional files].
References
Moutsopoulos HM, Chused TM, Mann DL, Klippel JH, Fauci AS, Frank MM, et al. Sjögren’s syndrome (Sicca syndrome): current issues. Ann Intern Med. 1980 Feb;92(2 Pt 1):212–26. https://doi.org/10.7326/0003-4819-92-2-212.
Skopouli FN, Dafni U, Ioannidis JP, Moutsopoulos HM. Clinical evolution, and morbidity and mortality of primary Sjögren’s syndrome. Semin Arthritis Rheum. 2000 Apr;29(5):296–304. https://doi.org/10.1016/s0049-0172(00)80016-5.
Beydon M, McCoy S, Nguyen Y, Sumida T, Mariette X, Seror R. Epidemiology of Sjögren syndrome. Nat Rev Rheumatol. 2024 Mar;20(3):158–69. https://doi.org/10.1038/s41584-023-01057-6.
Valim V, Zandonade E, Pereira AM, de Brito Filho OH, Serrano EV, Musso C, et al. Primary Sjögren’s syndrome prevalence in a major metropolitan area in Brazil. Rev Bras Reumatol. 2013 Feb;53(1):24–34.
Brito-Zerón P, Baldini C, Bootsma H, Bowman SJ, Jonsson R, Mariette X, et al. Sjögren syndrome. Nat Rev Dis Primers. Jul 2016;2:16047. https://doi.org/10.1038/nrdp.2016.47.
Seror R, Ravaud P, Bowman SJ, Baron G, Tzioufas A, Theander E, EULAR Sjögren’s Task Force, et al. EULAR Sjogren’s syndrome disease activity index: development of a consensus systemic disease activity index for primary Sjogren’s syndrome. Ann Rheum Dis. 2010 Jun;69(6):1103–09. https://doi.org/10.1136/ard.2009.110619. Epub 2009 Jun 28. Erratum in: Ann Rheum Dis. 2011 May;70(5):880.
Trevisani VFM, Pugliesi A, Pasoto SG, Lopes MLL, Guedes LKN, Miyamoto ST, et al. Recommendations for evaluation and diagnosis of extra-glandular manifestations of primary sjogren syndrome: results of an epidemiologic systematic review/meta-analysis and a consensus guideline from the Brazilian Society of Rheumatology (articular, pulmonary and renal). Adv Rheumatol. 2022 Jun;62(1):18. https://doi.org/10.1186/s42358-022-00248-1.
Trevisani VFM, Pinheiro AC, de Magalhães Souza Fialho SC, Fernandes MLMS, Pugliesi A, Pasoto SG, et al. Recommendations for evaluation and diagnosis of extra-glandular manifestations of primary Sjögren syndrome: results of an epidemiologic systematic review/meta-analysis and a consensus guideline from the Brazilian society of rheumatology (hepatic, gastrointestinal and pancreatic). Adv Rheumatol. 2022 Oct;62(1):35. https://doi.org/10.1186/s42358-022-00267-y.
Shiboski SC, Shiboski CH, Criswell L, Baer A, Challacombe S, Lanfranchi H, Sjögren’s International Collaborative Clinical Alliance (SICCA) Research Groups, et al. American College of Rheumatology classification criteria for Sjögren’s syndrome: a data-driven, expert consensus approach in the Sjögren’s International Collaborative Clinical Alliance cohort. Arthritis Care Res (Hoboken). 2012 Apr;64(4):475–87. https://doi.org/10.1002/acr.21591.
Vitali C, Bombardieri S, Jonsson R, Moutsopoulos HM, Alexander EL, Carsons SE, European Study Group on Classification Criteria for Sjögren’s Syndrome, et al. Classification criteria for Sjögren’s syndrome: a revised version of the European criteria proposed by the American-European Consensus Group. Ann Rheum Dis. 2002 Jun;61(6):554–58. https://doi.org/10.1136/ard.61.6.554.
Shiboski CH, Shiboski SC, Seror R, Criswell LA, Labetoulle M, Lietman TM, International Sjögren’s Syndrome Criteria Working Group, et al. 2016 American College of Rheumatology/European League against rheumatism classification criteria for primary Sjögren’s syndrome: a consensus and data-driven methodology involving three international patient cohorts. Arthritis Rheumatol. 2017 Jan;69(1):35–45. https://doi.org/10.1002/art.39859.
Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al. Cochrane handbook for systematic reviews of interventions. Wiley; 2019. https://doi.org/10.1002/9781119536604.
Munn Z, Moola S, Riitano D, Lisy K. The development of a critical appraisal tool for use in systematic reviews addressing questions of prevalence. Int J Health Policy Manag. 2014 Aug;3(3):123–28. https://doi.org/10.15171/ijhpm.2014.71.
Generali E, Costanzo A, Mainetti C, Selmi C. Cutaneous and mucosal manifestations of Sjögren’s syndrome. Clin Rev Allergy Immunol. 2017 Dec;53(3):357–70. https://doi.org/10.1007/s12016-017-8639-y.
Xuan L, Zhang YD, Li L, Zeng YP, Zhang HZ, Wang J, et al. Clinical profile and significance of mucocutaneous lesions of primary Sjögren’s syndrome: a large cross-sectional study with 874 patients. Chin Med J (Engl). 2017 Oct;130(20):2423–28. https://doi.org/10.4103/0366-6999.216403.
Walling H, Sontheimer R. Non-vasculitic cutaneous involvement. In: Ramos-Casals M, Stone J, Moutsopoulus H, editors. Sjögren’s Syndrome: diagnosis and Therapeutics. London: Springer-Verlag; 2012. p. 157–72.
Kittridge A, Routhouska SB, Korman NJ. Dermatologic manifestations of Sjögren syndrome. J Cutan Med Surg. 2011 Jan–Feb;15(1):8–14. https://doi.org/10.2310/7750.2010.09033.
Bernacchi E, Bianchi B, Amato L, Giorgini S, Fabbri P, Tavoni A, et al. Xerosis in primary Sjögren syndrome: immunohistochemical and functional investigations. J Dermatol Sci. 2005 Jul;39(1):53–55. https://doi.org/10.1016/j.jdermsci.2005.01.017.
Ueki H, Inagaki Y, Hamasaki Y, Ono M. Dermatologische Manifestationen des Sjögren-Syndroms [Dermatological manifestations of Sjögren’s syndrome. Hautarzt. 1991 Dec;42(12):741–47. (German).
Katayama I, Yokozeki H, Nishioka K. Impaired sweating as an exocrine manifestation in Sjögren’s syndrome. Br J Dermatol. 1995 Nov;133(5):716–20. https://doi.org/10.1111/j.1365-2133.1995.tb02744.x.
Engelke M, Jensen JM, Ekanayake-Mudiyanselage S, Proksch E. Effects of xerosis and ageing on epidermal proliferation and differentiation. Br J Dermatol. 1997 Aug;137(2):219–25. https://doi.org/10.1046/j.1365-2133.1997.18091892.x.
Provost TT, Watson R. Cutaneous manifestations of Sjögren’s syndrome. Rheum Dis Clin North Am. 1992;18:609–16. https://doi.org/10.1016/S0889-857X(21)00319-7.
Bernacchi E, Amato L, Parodi A, Cottoni F, Rubegni P, De Pità O, et al. Sjögren’s syndrome: a retrospective review of the cutaneous features of 93 patients by the Italian Group of Immunodermatology. Clin Exp Rheumatol. 2004;22.
Katayama I, Koyano T, Nishioka K. Prevalence of eyelid dermatitis in primary Sjögren’s syndrome. Int J Dermatol. 1994;33:421–24. https://doi.org/10.1111/j.1365-4362.1994.tb04044.x.
Katayama I, Teramoto N, Arai H, Nishioka K, Nishiyama S. Annular erythema. A comparative study of Sjögren syndrome with subacute cutaneous lupus erythematosus. Int J Dermatol. 1991 Sep;30(9):635–39. https://doi.org/10.1111/j.1365-4362.1991.tb03489.x.
Haimowitz JE, McCauliffe DP, Seykora J, Werth VP. Annular erythema of Sjögren’s syndrome in a white woman. J Am Acad Dermatol. 2000;42:1069–73. https://doi.org/10.1067/mjd.2000.105508.
Brito-Zerón P, Retamozo S, Akasbi M, Gandía M, Perez-De-Lis M, Soto-Cardenas MJ, et al. Annular erythema in primary Sjogren’s syndrome: description of 43 non-Asian cases. Lupus. 2014 Feb;23(2):166–75. https://doi.org/10.1177/0961203313515764.
Hsu S, Le EH, Khoshevis MR. Differential diagnosis of annular lesions. Am Fam Physician. 2001;64.
Ramos-Casals M, Brito-Zerón P, Bové A, Sisó-Almirall A. Sjögren’s syndrome: beyond sicca involvement. In: Khamasha M, Ramos-Casals M, editors. Autoimmune Diseases: acute and Complex Situations. London: Springer-Verlag; 2011. p. 45–66.
Seror R, Ravaud P, Mariette X, Bootsma H, Theander E, Hansen A, EULAR Sjögren’s Task Force, et al. EULAR Sjogren’s Syndrome Patient Reported Index (ESSPRI): development of a consensus patient index for primary Sjogren’s syndrome. Ann Rheum Dis. 2011 Jun;70(6):968–72. https://doi.org/10.1136/ard.2010.143743.
Seror R, Theander E, Bootsma H, Bowman SJ, Tzioufas A, Gottenberg JE, et al. Outcome measures for primary Sjögren’s syndrome: a comprehensive review. J Autoimmun. Jun 2014;51:51–56. https://doi.org/10.1016/j.jaut.2013.12.010.
Błochowiak K, Olewicz-Gawlik A, Polańska A, Nowak-Gabryel M, Kocięcki J, Witmanowski H, et al. Oral mucosal manifestations in primary and secondary Sjögren syndrome and dry mouth syndrome. Postepy Dermatol Alergol. 2016 Feb;33(1):23–27. https://doi.org/10.5114/pdia.2016.57764.
Alexander EL, Provost TT. Cutaneous manifestations of primary Sjögren’s syndrome: a reflection of vasculitis and association with anti-Ro(SSA) antibodies. J Invest Dermatol. 1983 May;80(5):386–91. https://doi.org/10.1111/1523-1747.ep12552002.
Ramos-Casals M, Anaya JM, García-Carrasco M, Rosas J, Bové A, Claver G, et al. Cutaneous vasculitis in primary Sjögren syndrome: classification and clinical significance of 52 patients. Medicine (Baltimore). 2004 Mar;83(2):96–106. https://doi.org/10.1097/01.md.0000119465.24818.98.
Demirkesen C. Approach to cutaneous vasculitides with special emphasis on small vessel vasculitis: histopathology and direct immunofluorescence. Curr Opin Rheumatol. 2017 Jan;29(1):39–44. https://doi.org/10.1097/BOR.0000000000000346.
Both T, Dalm VA, van Hagen PM, van Daele PL. Reviewing primary Sjögren’s syndrome: beyond the dryness - from pathophysiology to diagnosis and treatment. Int J Med Sci. 2017 Feb;14(3):191–200. https://doi.org/10.7150/ijms.17718.
Ramos-Casals M, Brito-Zerón P, Yagüe J, Akasbi M, Bautista R, Ruano M, et al. Hypocomplementaemia as an immunological marker of morbidity and mortality in patients with primary Sjogren’s syndrome. Rheumatology (Oxford). 2005 Jan;44(1):89–94. https://doi.org/10.1093/rheumatology/keh407.
Gemignani F, Marbini A, Pavesi G, Di Vittorio S, Manganelli P, Cenacchi G, et al. Peripheral neuropathy associated with primary Sjogren’s syndrome. J Neurol Neurosurg Psychiatry. 1994;57:983–86.
Tzioufas AG, Boumba DS, Skopouli FN, Moutsopoulos HM. Mixed monoclonal cryoglobulinemia and monoclonal rheumatoid factor cross-reactive idiotypes as predictive factors for the development of lymphoma in primary Sjögren’s syndrome. Arthritis Rheum. 1996;39:767–72. https://doi.org/10.1002/art.1780390508.
Voulgarelis M, Tzioufas AG, Moutsopoulos HM. Mortality in Sjögren’s syndrome. Clin Exp Rheumatol. 2008 Sep–Oct;26(5 Suppl 51):S66–71.
Meijer JM, Schonland SO, Palladini G, Merlini G, Hegenbart U, Ciocca O, et al. Sjögren’s syndrome and localized nodular cutaneous amyloidosis: coincidence or a distinct clinical entity? Arthritis Rheum. 2008 Jul;58(7):1992–99. https://doi.org/10.1002/art.23617.
Yamamoto T, Ohi M, Nishioka K. Lymphomatoid papulosis associated with Sjögren’s syndrome. J Dermatol. 2002 Mar;29(3):174–77. https://doi.org/10.1111/j.1346-8138.2002.tb00244.x.
Tsuboi H, Katsuoka K. Ulcerative lichen planus associated with Sjögren’s syndrome. J Dermatol. 2007 Feb;34(2):131–34. https://doi.org/10.1111/j.1346-8138.2006.00232.x.
Tsuruta D, Matsumura-Oura A, Ishii M. Subcorneal pustular dermatosis and Sjögren’s syndrome. Int J Dermatol. 2005 Nov;44(11):955–57. https://doi.org/10.1111/j.1365-4632.2004.02290.x.
Aoki A, Ohno S, Ueda A, Ideguchi H, Ohkubo T, Hagiwara E, et al. [Hematological abnormalities of primary Sjogren’s syndrome]. Nihon Rinsho Meneki Gakkai Kaishi. 2000 Apr;23(2):124–28 (Japanese). https://doi.org/10.2177/jsci.23.124.
Brito-Zerón P, Soria N, Muñoz S, Bové A, Akasbi M, Belenguer R, et al. Prevalence and clinical relevance of autoimmune neutropenia in patients with primary Sjögren’s syndrome. Semin Arthritis Rheum. 2009 Apr;38(5):389–95. https://doi.org/10.1016/j.semarthrit.2008.01.014.
Ramos-Casals M, Solans R, Rosas J, Camps MT, Gil A, Del Pino-Montes J, GEMESS Study Group, et al. Primary Sjögren syndrome in Spain: clinical and immunologic expression in 1010 patients. Medicine (Baltimore). 2008 Jul;87(4):210–19. https://doi.org/10.1097/MD.0b013e318181e6af.
Baldini C, Pepe P, Quartuccio L, Priori R, Bartoloni E, Alunno A, et al. Primary Sjogren’s syndrome as a multi-organ disease: impact of the serological profile on the clinical presentation of the disease in a large cohort of Italian patients. Rheumatology (Oxford). 2014 May;53(5):839–44. https://doi.org/10.1093/rheumatology/ket427.
Ramos-Casals M, Brito-Zerón P, Solans R, Camps MT, Casanovas A, Sopeña B, SS Study Group; Autoimmune Diseases Study Group (GEAS) of the Spanish Society of Internal Medicine (SEMI), et al. Systemic involvement in primary Sjogren’s syndrome evaluated by the EULAR-SS disease activity index: analysis of 921 Spanish patients (GEAS-SS Registry). Rheumatology (Oxford). 2014 Feb;53(2):321–31. https://doi.org/10.1093/rheumatology/ket349.
Friedman JA, Miller EB, Green L, Huszar M, Schattner A. A community-based cohort of 201 consecutive patients with primary Sjögren’s syndrome in Israel: ashkenazi patients compared with those of Sephardic descent. Clin Exp Rheumatol. 2006;24.
Barcelos F, Abreu I, Patto JV, Trindade H, Teixeira A. Anti-cyclic citrullinated peptide antibodies and rheumatoid factor in Sjögren’s syndrome. Acta Reumatol Port. 2009;34.
Baimpa E, Dahabreh IJ, Voulgarelis M, Moutsopoulos HM. Hematologic manifestations and predictors of lymphoma development in primary Sjögren syndrome: clinical and pathophysiologic aspects. Medicine (Baltimore). 2009 Sep;88(5):284–93. https://doi.org/10.1097/MD.0b013e3181b76ab5.
Zhou JG, Qing YF, Jiang L, Yang QB, Luo WF. Clinical analysis of primary Sjögren’s syndrome complicating anemia. Clin Rheumatol. 2010 May;29(5):525–29. https://doi.org/10.1007/s10067-009-1366-x.
Caramaschi P, Biasi D, Caimmi C, Scambi C, Pieropan S, Barausse G, et al. The co-occurrence of Hashimoto thyroiditis in primary Sjogren’s syndrome defines a subset of patients with milder clinical phenotype. Rheumatol Int. 2013 May;33(5):1271–75. https://doi.org/10.1007/s00296-012-2570-6.
Malladi AS, Sack KE, Shiboski SC, Shiboski CH, Baer AN, Banushree R, et al. Primary Sjögren’s syndrome as a systemic disease: a study of participants enrolled in an international Sjögren’s syndrome registry. Arthritis Care Res (Hoboken). 2012 Jun;64(6):911–18. https://doi.org/10.1002/acr.21610.
Retamozo S, Gheitasi H, Quartuccio L, Kostov B, Corazza L, Bové A, et al. Cryoglobulinaemic vasculitis at diagnosis predicts mortality in primary Sjögren syndrome: analysis of 515 patients. Rheumatology (Oxford). 2016 Aug;55(8):1443–51. https://doi.org/10.1093/rheumatology/kew194.
Xu D, Zhao S, Li Q, Wang YH, Zhao JL, Li MT, et al. Characteristics of Chinese patients with primary Sjögren’s syndrome: preliminary report of a multi-centre registration study. Lupus. 2020 Jan;29(1):45–51. https://doi.org/10.1177/0961203319889666.
Ramakrishna R, Chaudhuri K, Sturgess A, Manoharan A. Haematological manifestations of primary Sjögren’s syndrome: a clinicopathological study. QJM. 1992;83. https://doi.org/10.1093/oxfordjournals.qjmed.a068692.
Liu Y, Chen S, Sun Y, Lin Q, Liao X, Zhang J, et al. Clinical characteristics of immune thrombocytopenia associated with autoimmune disease: a retrospective study. Medicine (Baltimore). 2016 Dec;95(50):e5565. https://doi.org/10.1097/MD.0000000000005565.
Theander E, Manthorpe R, Jacobsson LT. Mortality and causes of death in primary Sjögren’s syndrome: a prospective cohort study. Arthritis Rheum. 2004 Apr;50(4):1262–69. https://doi.org/10.1002/art.20176.
Ioannidis JP, Vassiliou VA, Moutsopoulos HM. Long-term risk of mortality and lymphoproliferative disease and predictive classification of primary Sjögren’s syndrome. Arthritis Rheum. 2002 Mar;46(3):741–47. https://doi.org/10.1002/art.10221.
Quartuccio L, Baldini C, Priori R, Bartoloni E, Carubbi F, Alunno A, GRISS Group, et al. Cryoglobulinemia in Sjögren syndrome: a disease subset that links higher systemic disease activity, autoimmunity, and local B cell proliferation in mucosa-associated lymphoid tissue. J Rheumatol. 2017 Aug;44(8):1179–83. https://doi.org/10.3899/jrheum.161465.
Brito-Zerón P, Acar-Denizli N, Zeher M, Rasmussen A, Seror R, Theander E, EULAR-SS Task Force Big Data Consortium, et al. Influence of geolocation and ethnicity on the phenotypic expression of primary Sjögren’s syndrome at diagnosis in 8310 patients: a cross-sectional study from the Big Data Sjögren Project Consortium. Ann Rheum Dis. 2017 Jun;76(6):1042–50. https://doi.org/10.1136/annrheumdis-2016-209952.
Lin DF, Yan SM, Zhao Y, Zhang W, Li MT, Zeng XF, et al. Clinical and prognostic characteristics of 573 cases of primary Sjögren’s syndrome. Chin Med J (Engl). 2010:123. https://doi.org/10.3760/cma.j.issn.0366-6999.2010.22.015.
Fauchais AL, Ouattara B, Gondran G, Lalloué F, Petit D, Ly K, et al. Articular manifestations in primary Sjögren’s syndrome: clinical significance and prognosis of 188 patients. Rheumatology (Oxford). 2010 Jun;49(6):1164–72. https://doi.org/10.1093/rheumatology/keq047.
Risselada AP, Kruize AA, Bijlsma JW. Clinical features distinguishing lymphoma development in primary Sjögren’s syndrome-a retrospective cohort study. Semin Arthritis Rheum. 2013 Oct;43(2):171–77. https://doi.org/10.1016/j.semarthrit.2013.03.001.
Alamanos Y, Tsifetaki N, Voulgari PV, Venetsanopoulou AI, Siozos C, Drosos AA. Epidemiology of primary Sjögren’s syndrome in north-west Greece, 1982-2003. Rheumatology (Oxford). 2006 Feb;45(2):187–91. https://doi.org/10.1093/rheumatology/kei107.
Ramos-Casals M, Cervera R, Yagüe J, García-Carrasco M, Trejo O, Jiménez S, et al. Cryoglobulinemia in primary Sjögren’s syndrome: prevalence and clinical characteristics in a series of 115 patients. Semin Arthritis Rheum. 1998 Dec;28(3):200–05. https://doi.org/10.1016/s0049-0172(98)80037-1.
García-Carrasco M, Ramos-Casals M, Rosas J, Pallarés L, Calvo-Alen J, Cervera R, et al. Primary Sjögren syndrome: clinical and immunologic disease patterns in a cohort of 400 patients. Medicine (Baltimore). 2002 Jul;81(4):270–80. https://doi.org/10.1097/00005792-200207000-00003.
Vasil’ev VI, Khodarev NV, Mach ES, Manuĭlova LS, Simonova MV, Svobodina ON, et al. Krioglobulinemiia pri bolezni Shegrena [Cryoglobulinemia in Sjögren’s syndrome. Ter Arkh. 1990;62(5):66–70 (Russian).
Vasil’ev VI, Probatova NA, Varlamova E, Tupitsin NN, Simonova MV, Safonova TN, et al. Prognosticheskoe znachenie smeshannoĭ monoclonal’noĭ krioglobulinemii pri bolezni Shegrena [Prognostic implications of mixed monoclonal cryoglobulinemia in Sjogren’s disease]. Ter Arkh. 2004;76(8):61–68 (Russian).
Ekström Smedby K, Vajdic CM, Falster M, Engels EA, Martínez-Maza O, Turner J, et al. Autoimmune disorders and risk of non-Hodgkin lymphoma subtypes: a pooled analysis within the InterLymph Consortium. Blood. 2008 Apr;111(8):4029–38. https://doi.org/10.1182/blood-2007-10-119974.
Papageorgiou A, Ziogas DC, Mavragani CP, Zintzaras E, Tzioufas AG, Moutsopoulos HM, et al. Predicting the outcome of Sjogren’s syndrome-associated non-hodgkin’s lymphoma patients. PLoS One. 2015 Feb;10(2):e0116189. https://doi.org/10.1371/journal.pone.0116189.
Goules AV, Tzioufas AG. Lymphomagenesis in Sjögren’s syndrome: predictive biomarkers towards precision medicine. Autoimmun Rev. 2019 Feb;18(2):137–43. https://doi.org/10.1016/j.autrev.2018.08.007.
Kiesewetter B, Lamm W, Dolak W, Lukas J, Mayerhoefer ME, Weber M, et al. Transformed mucosa-associated lymphoid tissue lymphomas: a single institution retrospective study including polymerase chain reaction-based clonality analysis. Br J Haematol. 2019 Aug;186(3):448–59. https://doi.org/10.1111/bjh.15953.
Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016 May;127(20):2375–90. https://doi.org/10.1182/blood-2016-01-643569.
Nishishinya MB, Pereda CA, Muñoz-Fernández S, Pego-Reigosa JM, Rúa-Figueroa I, Andreu JL, et al. Identification of lymphoma predictors in patients with primary Sjögren’s syndrome: a systematic literature review and meta-analysis. Rheumatol Int. 2015 Jan;35(1):17–26. https://doi.org/10.1007/s00296-014-3051-x.
Fragkioudaki S, Mavragani CP, Moutsopoulos HM. Predicting the risk for lymphoma development in Sjogren syndrome: an easy tool for clinical use. Medicine (Baltimore). 2016 Jun;95(25):e3766. https://doi.org/10.1097/MD.0000000000003766.
Feinstein AJ, Ciarleglio MM, Cong X, Otremba MD, Judson BL. Parotid gland lymphoma: prognostic analysis of 2140 patients. Laryngoscope. 2013 May;123(5):1199–203. https://doi.org/10.1002/lary.23901.
Zucca E, Bertoni F. The spectrum of MALT lymphoma at different sites: biological and therapeutic relevance. Blood. 2016 Apr;127(17):2082–92. https://doi.org/10.1182/blood-2015-12-624304.
Kws K, Bhatia KS, Qyh A, King AD. Imaging of head and neck mucosa-associated lymphoid tissue lymphoma (MALToma). Cancer Imaging. 2021 Jan;21(1):10. https://doi.org/10.1186/s40644-020-00380-5.
Acknowledgements
Brazilian Society of Rheumatology. To the most recent members of the sjogren’s disease committee: Carolina Yume Arazawa, Erica Vieira Serrano, Erika R.S. Batista Mendonça, Michelle Alencar Pereira, Samia Araujo de Souza Studart, Sandra Rejane Cabral Batista.
Funding
The authors have no financial sponsors for this study.
Author information
Authors and Affiliations
Contributions
All the authors contributed equally to this work. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
AP: None; DCSE: None; DUC: None; GD: None; SGP: None; FRO: None; VV: None MLLL: None; STM: None; MLMSF: None; SCMSF: None; ACP: None; LCS: None; SA: None; SLER: None; TNLK: None; MCLFSS: None; JDG: None; RP: None; KGC: None;. VTC: None; ACPNP: None; CRRF: None; APR: None; VFMT: None.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This paper is a publication by the Sj?gren?s Disease Commission of the Brazilian Society of Rheumatology.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pugliesi, A., Egypto, D., Duffles, G. et al. Recommendations on cutaneous and hematological manifestations of Sjögren’s disease by the Brazilian Society of Rheumatology. Adv Rheumatol 64, 51 (2024). https://doi.org/10.1186/s42358-024-00391-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42358-024-00391-x