
Abstract
Aim
This study aimed to investigate the causal impact of inflammatory cytokines on Sjogren’s Syndrome (SS) and to identify potential biomarkers for SS clinical management using Mendelian Randomization (MR).
Materials and methods
Leveraging GWAS summary data of inflammatory cytokines and SS, we executed the first two-sample MR analysis. Genetic variants from prior GWASs associated with circulating inflammatory cytokines served as instrumental variables (IVs). Data regarding cytokines were analyzed using the Olink Target-96 Inflammation panel, synthesizing data from 14,824 participants. GWAS summary statistics for SS were procured from the UK Biobank, focusing on samples of European ancestry. To discern the causal relationship between inflammatory cytokines and SS, several MR methodologies, including inverse variance weighted (IVW) and MR-Egger regression, were applied.
Results
After rigorous IV quality control, 91 cytokines were incorporated into the MR analysis. The IVW analysis identified 8 cytokines with a positive association to SS: Axin-1 (OR 2.56, 95% CI 1.07–6.10), T-cell surface glycoprotein CD5 (OR 1.81, 95% CI 1.08–3.02), CUDP1 (OR 1.61, 95% CI 1.00-2.58), CXCL10 (OR 1.92, 95% CI 1.25–2.95), IL-4 (OR 2.18, 95% CI 1.22–3.91), IL-7 (OR 2.35, 95% CI 1.27–4.33), MCP-2 (OR 1.27, 95% CI 1.05–1.54), and TNFRSF9 (OR 1.83, 95% CI 1.03–3.24), suggesting their potential in increasing SS risk.
Conclusion
Our study conducted through MR, identified various inflammatory cytokines associated with SS risk, validating some previous research results and offering some new potential biomarkers for SS. However, these findings necessitate further research for validation and exploration of their precise role in the onset and progression of SS.
Similar content being viewed by others


Introduction
Sjogren’s Syndrome (SS), a prevalent autoimmune epithelitis, primarily targets the lacrimal and salivary glands, resulting in conditions such as xerophthalmia and xerostomia [1, 2]. The disease manifests either as primary SS, a standalone condition, or secondary SS, where it associates with other connective tissue diseases [2]. The pathological hallmark of SS is the infiltration of lymphocytes, mainly CD4 + T cells, into the secretory organs [3]. This disease has been implicated in a range of systemic symptoms, such as fever, arthralgia, and long-term fatigue, further complicating its clinical presentation [4].
Understanding the pathogenesis of SS is a complex undertaking, involving a myriad of cellular and molecular players [2, 5]. Among these, cytokines have emerged as critical factors in the ongoing debate about the disease’s etiology [6]. Initially, the immune response in SS was largely attributed to T cells, particularly the T helper cells (Th), categorized into Th1 and Th2 based on their cytokine profile [7]. However, the advent of newer subsets like Th17 cells and the acknowledgment of cytokine-producing B cells have added layers of complexity to this already intricate landscape [8, 9]. In the early stages of SS, lymphocytes infiltrate the affected glands, setting the stage for an inflammatory cascade [10]. Cytokines such as interleukin (IL)-17 produced by Th17 cells and others like interferon (IFN)-γ play a central role in mediating this inflammation. As the disease progresses, the balance of these cytokines can shift, influencing the severity and the type of symptoms experienced by the patient [11]. Despite strides in understanding SS, numerous enigmas endure. The precise role of cytokines in manifesting SS’s clinical symptoms, and the extent to which they underpin systemic symptoms, remains shrouded [12]. The inadequacy of T-cell-centric therapies has cast doubts on the predominant role of T cells in SS, thereby pivoting the spotlight towards B cells and their cytokine profiles [13]. Persistent research voids, fueled by inconsistent findings often stemming from constrained sample sizes or study design inadequacies, beckon further inquiry to unfurl the mystifying immunological tapestry of SS.
Mendelian Randomization (MR) is emerging as a powerful analytical tool for making sense of the intricate web of factors that contribute to diseases. Utilizing genetic variations associated with exposures, MR can act like a “genetic detective” to infer potential causal relationships between these exposures and observed health outcomes [14, 15]. It is often compared to a “natural” randomized controlled trial (RCT), as it exploits the random distribution of alleles during gamete production [16, 17]. This process is like Mother Nature’s own version of a blind draw, effectively minimizing confounding variables and biases, including those resulting from reverse causality [18]. In recent years, MR studies have shed new light on the complex interplay between autoimmune diseases and neurodegenerative conditions, such as Alzheimer’s Disease [19]. Specifically, this research has suggested that liability to autoimmune diseases like multiple sclerosis and SS may be associated with Alzheimer’s Disease, although the mechanisms behind these associations warrant further exploration. At the same time, MR studies have looked into other factors related to SS. One study focused on how gut bacteria could affect the risk of getting SS [20]. Some bacteria were found to increase the risk, while others reduced it. Another study checked if vitamin D levels had any effect on SS but didn’t find a clear link [21].
In this study, we performed the first two-sample MR analysis of the Genome-Wide Association Studies (GWASs) summary data containing inflammatory cytokines and SS, revealed the causal impact of inflammatory cytokines on SS, provided new biomarkers for the clinical management of SS.
Materials and methods
Study design
The MR study conducted adhered to three core instrumental variable (IV) assumptions. (1) the genetic variants selected must exhibit a correlation with the exposure. (2) the chosen variants should remain free from any confounding factors. (3) the variants should only influence the outcome through the exposure (Fig. 1). Data summaries pertaining to circulating inflammatory cytokines and SS were acquired from publicly accessible GWASs, with a primary focus on cohorts of European descent as cited by reference [8]. The methodology of this MR study was aligned with the guidelines stipulated by Strengthening the Reporting of Observational Studies in Epidemiology using MR (STROBE-MR).

Diagram of the MR analysis. Assumption 1, genetic instruments are strongly associated with the exposures of interest; Assumption 2, genetic instruments are independent of confounding factors; Assumption 3, genetic instruments are not associated with outcome and affect outcome only via exposures. IVW, inverse variance weighted; LD, linkage disequilibrium; LOO analysis, leave-one-out analysis; WMedine, weighted medine; SNPs, single nucleotide polymorphisms; WM, weighted mode
Circulating inflammatory cytokines data source
GWAS summary statistics for circulating inflammatory cytokines were obtained from an up-to-date research launched by the SCALLOP Consortium [22]. The study was designed to explore inflammatory responses resulting in tissue damage and are central to the pathogenesis of multiple diseases, including sepsis, autoimmunity and atherothrombosis. Cytokines were analyzed using the Olink Target-96 Inflammation panel, with 91 proteins included due to BDNF issues [22]. Key or potential cytokines such as IL-4 [23], IL-6 [24], IL-7 [25], IL-10 [26], IL-12 [27], IL-15 [28], IL-17 [29], IL-22 [30], IL-33 [31], C-X-C motif chemokine 10 (CXCL10) [32], etc. for Sjogren’s disease research were included in the study (Table S1). The data was generated at Olink’s labs in Uppsala. Genotyping used SNP arrays, with imputation via 1000 Genomes or HRC panel. A GWAS analysis was employed for each protein in the pQTL mapping. The meta-analysis synthesized data from 14,824 participants, setting a statistical significance at P ≤ 5 × 10− 10 [22]. Results were compared to the ARISTOTLE study [33, 34]. pQTLs, reflecting protein abundance associations, were clearly defined. Protein variance was computed through a formula, and conditional analysis utilized GCTA [22].
SS data source
GWAS summary statistics for SS were obtained from the UK Biobank [35]. The study involved approximately 500,000 participants aged 40–69, with extensive phenotypic information recorded. Genotyping was executed with the Affymetrix UK BiLEVE Axiom array for the first 50,000 and the Affymetrix UK Biobank Axiom for the rest. Analysis used up to 11,914,699 variants from the Haplotype Reference Consortium panel, focusing on samples of European ancestry. After selecting 407,746 individuals of white British ancestry and applying quality filters via PLINK2 [36] that included: a minor allele frequency of ≥ 1%, a Hardy–Weinberg equilibrium test not exceeding P = 1 × 10− 15, a genotyping rate above 99%, not present in low-complexity regions, not involved in inter-chromosomal LD and LD pruning using a R2 threshold of 0.9 with a window size of 1,000 markers and a step size of 100 markers, up to 471,762 genotyped SNPs were retained for further analysis [35]. The SS data utilized in this study encompass both primary and secondary SS.
Selection of IVs
Single nucleotide polymorphism (SNP)s from prior GWASs pertinent to circulating inflammatory cytokines were employed for MR assessment, adhering to a genome-wide significance threshold (p < 5 × 10− 6)). The chosen SNPs ensured no linkage disequilibrium (LD) with other SNPs, maintaining an \( {r}^{2}\) below 0.1 within a 500 kb clumping radius. Where SNPs surpassed the \( {r}^{2}\) = 0.1 threshold, only the SNP with the strongest association (smallest P value) with the cytokine was chosen. This selection approach aligns with methods traditionally utilized in preceding studies [37, 38]. To counter possible bias from subpar instruments, the \( {R}^{2}\) and F statistics for each SNP were ascertained using specified formulas:
Here, β, EAF, se(β), N, and k denote genetic variant effect size, effect allele frequency, standard error of this effect size, exposure sample size, and SNP count, respectively. SNPs with F below 10 were discarded. Procedures extracted and harmonized outcome-associated SNPs, excluding correlated (p < 5 × 10− 6), palindromic, and allele inconsistent SNPs, leading to an MR study on cytokines with over two SNPs [39].
Statistical analysis
To ascertain the causal relationship between circulating inflammatory cytokines and SS, a multi-faceted methodological approach was adopted in this research. This included the utilization of inverse variance weighted (IVW), MR-Egger regression, MR-Egger intercept, weighted median and weighted mode strategies. The aggregate effect of circulating inflammatory cytokines on SS was delineated via a meta-analysis technique, amalgamating Wald estimates for each SNP through the IVW method [17]. In endorsing significant results, heterogeneity and horizontal pleiotropy tests were conducted employing meta-analytical techniques, encompassing the modified Cochran Q statistic calculated for IVW and MR-Egger estimates [40]. To mitigate the heterogeneity impact attributed to a singular SNP, a leave-one-out analysis was enacted, systematically excluding one SNP at a time. Under the absence of horizontal pleiotropy, IVW results remain impartial. The MR-Egger regression, predicated on the InSIDE assumption that instrument strength bears no correlation to a direct effect, evaluates pleiotropy through its intercept term. A zero intercept term in x aligns with IVW results, denoting an absence of horizontal pleiotropy [14]. The weighted median technique facilitates accurate causal deductions, even with up to 50% of the IVs deemed invalid [31]. When the InSIDE assumption faces challenges, the weighted mode estimate yields augmented power, diminished bias, and a reduced type I error rate for MR-Egger regression [41]. The evaluations were facilitated by the “TwoSampleMR” R packages, version 4.1.3 [42].
Result
Selection of instrumental variables
Upon stringent quality control of IVs, 91 cytokines were included in the MR analysis (Supplementary Table S1). These IVs comprised SNPs ranging from 9 to 27 (with Axin-1 genetically represented by 9 SNPs and tumor necrosis factor receptor superfamily member 9 levels having the highest representation with 27 SNPs). No evidence of pleiotropic effects was detected by the MR regression intercept test (P > 0.05). The F-statistics of IVs ranged between 20.90 and 4995.16, all largely > 10, indicating no evidence of weak instrument bias (Table S2).
Causal effects of cytokines on SS
Subsequent IVW analysis, paired with supplemental and sensitivity evaluations, pinpointed 8 cytokines that satisfied the stringent selection criteria as potential candidates (Fig. 2) (Table 1). The selected risk factors were as followed: Axin-1 levels, T-cell surface glycoprotein CD5 (T-cell CD5) levels, CUB domain-containing protein 1 (CUDP1) levels, CXCL10 levels, IL-4 levels, IL-7 levels, Monocyte chemoattractant protein 2 (MCP-2) levels, Tumor necrosis factor receptor superfamily member 9 (TNFRSF9) levels. All selected cytokines are positive association with SS: Axin-1 levels (OR 2.56, 95% CI 1.07 to 6.10, p = 0.034), T-cell CD5 levels (OR 1.81, 95% CI 1.08 to 3.02, p = 0.024), CUDP1 levels (OR 1.61, 95% CI 1.00 to 2.58, p = 0.049), CXCL10 levels (OR 1.92, 95% CI 1.25 to 2.95, p = 0.003), IL-4 levels (OR 2.18, 95% CI 1.22 to 3.91, p = 0.009), IL-7 levels (OR 2.35, 95% CI 1.27 to 4.33, p = 0.006), MCP-2 levels (OR 1.27, 95% CI 1.05 to 1.54, p = 0.011), TNFRSF9 (OR 1.83, 95% CI 1.03 to 3.24, p = 0.040). This suggests that these cytokines may increase the risk of SS.

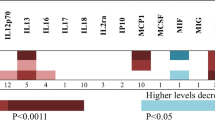
Forest plot of MR analysis. Forest plot to visualize the causal effect of cytokines on the risk of SS risk factors by Inverse variance weighted, MR Egger regression, Weighted median and Weighted mode method. Axin-1 levels, T-cell surface glycoprotein CD5 levels, CUDP1 levels, CXCL10 levels, IL-4 levels, IL-7 levels, MCP-2 levels and TNFRSF9. This plot shows these cytokines may increase the risk of SS.
In summation, IVW-derived estimates were significant (p < 0.05), and there was consistency in direction and magnitude across IVW, MR-Egger, weighted median, and weighted mode estimates (Fig. 3) (Supplementary Table S3). Scatter plots for identified cytokines across various tests are displayed on Fig. 4. Both the Cochran Q test (p > 0.05) and the MR-Egger intercept test (p > 0.05) strongly supported the lack of heterogeneity and pleiotropy (Table 2). Leave-one-out analysis affirmed that no individual SNP introduced bias into the MR estimation (SupplementaryFigure S1). The funnel plots were showed on SupplementaryFigure S2.

Preliminary MR analyses for the associations between inflammatory cytokines and the risk of SS. The circle from the outer to the inner represented the IVW, MR-Egger regression, weighted median, and weighted mode, respectively. The shades of color were reflections of the magnitude of the p-value as the label inside the circle. (MR Egger, MR Egger regression; IVW, inverse variance-weighted.)

Scatter plots of MR analysis. Analyses were conducted using IVW, MR Egger, Weighted median and Weighted mode. The slope of each line corresponding to the estimated MR effect per method
Discussion
SS is an autoimmune condition that primarily affects the salivary and lacrimal glands, leading to conditions like xerostomia (dry mouth) and xerophthalmia (dry eyes). The intricate dance of cytokines, receptors, and immune cells contributes to the onset and progression of this disease. Our MR experiment has shed light on several key players in this scenario, and integrating the findings from various studies paints a richer picture of the SS.
The results indicate a strong association between increased levels of CXCL10 and the risk of an autoimmune response (OR 1.92, p = 0.003). The chemokine CXCL10 is known to exert its functions through its receptor CXCR3 [43]. This binding plays a pivotal role in the pathogenesis of a myriad of autoimmune diseases, ranging from organ-specific diseases such as type 1 diabetes and Graves’ disease to systemic conditions like rheumatoid arthritis and SS [44]. A mechanism through which this occurs is that the secretion of CXCL10 by various immune cells, including CD4+, CD8+, NK, and NK-T cells, is dependent on IFN-γ, which itself is mediated by the interleukin-12 cytokine family [45]. It is interesting to note that high levels of CXCL10 in peripheral fluids act as a marker of host immune response, predominantly by Th1 orientated T-cells [46]. This Th1 response in tissues potentially leads to the enhanced production of IFN-γ and tumor necrosis factor-α, stimulating further CXCL10 secretion [47]. This amplification feedback loop thus perpetuates the autoimmune process. Given these insights, CXCL10 could indeed be a novel therapeutic target for autoimmune diseases.
CD5, a T-cell surface glycoprotein, exhibited an association with increased autoimmune risk (OR 1.81, p = 0.024). Delving into the literature, CD5 seems to play an intriguing role in SS [48]. In patients with primary SS, a reduced expression and function of CD5 molecule on peripheral blood lymphocytes were observed [49]. Additionally, the ratio of CD5 + to CD3 + lymphocytes was significantly lower in these patients compared to normal subjects, highlighting a CD5 deficiency [48]. This reduction has potential implications, as CD5 could be involved in intracellular signaling defects in primary SS [49]. This conclusion is further solidified by observations where a correction in the CD5 lymphocyte abnormality was associated with clinical remission in some SS patients. Additionally, studies on lip biopsy specimens from SS patients also revealed that a significant portion of cellular infiltrates expressed the CD5 molecule [48, 50]. This further accentuates the importance of CD5 in the disease pathology of SS.
The role of IL-7, another interleukin, in SS cannot be understated. Our results show a noteworthy association between elevated Interleukin-7 levels and the disease (OR 2.35, p = 0.006). This is consistent with the findings from a single-center study, where primary SS patients exhibited higher serum IL-7 levels [25]. The systematic scoping review further emphasized the potential of IL-7 as a biomarker for monitoring primary SS activity [51]. Elevated IL-7 levels, mainly from salivary glands, could be pivotal in primary SS immunopathology. Transitioning to IL-4 (OR 2.18, p = 0.009), research in NOD/LtJ mice linked to SS found increased B cell infiltration and salivary gland apoptosis. Leptin treatment led to IL-4 secretion from B cells, hinting at the Leptin/OB-R pathway’s role in promoting SGEC apoptosis [52]. Studies using IL-4 gene knockout NOD mice highlighted IL-4’s essential role in autoimmune xerostomia development. While the exact mechanism remains uncertain, IL-4’s impact exists [53].
Our study also identified a significant association between elevated TNFRSF9 levels and SS (OR 1.83, p = 0.04). But there is no previous study discovered TNFRSF9’s relation to SS. However, CD137 (4-1BB), a surface glycoprotein that belongs to TNFRSF9 has reported having relation to SS. This finding is particularly noteworthy because 4-1BB is a costimulatory receptor that has been shown to modulate T cell responses. A study on the NOD model of SS highlighted that activation of 4-1BB could impede the development of sialadenitis by modulating various immune cell types and their associated cytokines [54, 55]. Previous studies in MRL-Faslpr mice also demonstrated that the expression of costimulatory molecules GITRL and 4-1BBL in salivary glands was significantly correlated with the severity of autoimmune sialadenitis [55]. Our findings are in line with these observations and extend them by providing genetic evidence may implicating TNFRSF9 in the pathogenesis of SS.
Our study also discovered new cytokine related to SS. Axin-1 levels, MCP-2 levels and CDCP1 levels have been found having a significant positive correlation between the levels of the risk of SS, which is not reported in previous studies. This may suggest a diverse role of the cytokine in the onset and progression of SS. The novel findings may contribute to a more comprehensive understanding of the circulating inflammatory cytokine of SS and offer new potential biomarkers for the early diagnosis and prevention of SS.
Several limitations exist within this study. Firstly, the exposure of interest at the genome-wide level had a restricted number of SNPs. This was addressed by applying slightly relaxed thresholds for the MR analysis, mirroring practices in previous research. Nonetheless, with F-statistic values for the chosen SNPs surpassing 10, the robustness of our IVs is indicated. Secondly, our MR analysis exclusively utilized GWAS data from European ancestry individuals, limiting ethnic variability. Consequently, the applicability of these findings to diverse populations necessitates further investigation and validation. Thirdly, the number of cytokines included in this study is limited. Prior genomic studies have identified associations in SS with molecules of the human leukocyte antigen complex and with transcription factors related to the IFN signature [56,57,58]. Therefore, analysis about broader array of cytokines is needed in the future. Fourthly, the SS data utilized in this study encompass both primary and secondary SS. Material from primary SS or secondary SS may influence genomic and proteomic analysis. Carballeda J et al.’s study reported primary SS patients exhibited a greater number of CD4(+)/IL-17 A(+) and IL-19(+) T cells but a lower percentage of IL-24(+) cells (P < 0.05) than secondary SS [59]. Subsequent analyses that categorize SS into these subtypes may uncover distinct impacts of cytokines on primary versus secondary SS. Fifthly, the MR estimation’s precision is inherently tied to sample size, emphasizing the need to augment the sample size for result validation. While MR analysis sheds light on disease etiology, it’s imperative to corroborate our findings through rigorous RCTs and foundational research before clinical integration.
Conclusion
our study, conducted through MR, identified various inflammatory cytokines associated with SS risk, validating some previous research results and offering some new potential biomarkers for SS. However, these findings necessitate further research for validation and exploration of their precise role in the onset and progression of SS.
Data availability
We have annotated the article with the source of all original data, please contact the original authors for access if needed. The results of this study can be obtained by contacting the corresponding author.
Abbreviations
- SS:
-
Sjogren’s Syndrome
- MR:
-
Mendelian Randomization
- RCT:
-
Randomized controlled trial
- IV:
-
Instrumental variable
- IVW:
-
Inverse variance weighted
- STROBE-MR:
-
Strengthening the Reporting of Observational Studies in Epidemiology using MR
- LD:
-
Linkage disequilibrium
- CUDP1:
-
CUB domain-containing protein 1
- CXCL10:
-
C-X-C motif chemokine 10
- IL:
-
Interleukin
- MCP-2:
-
Monocyte chemoattractant protein 2
- TNFRSF9:
-
Tumor necrosis factor receptor superfamily member 9 levels
- SNP:
-
Single nucleotide polymorphism
- IFN:
-
interferon
References
Skopouli FN, Moutsopoulos HM. Autoimmune epitheliitis: Sjögren’s syndrome. Clin Exp Rheumatol. 1994;12(Suppl 11):9–11.
Jonsson R, Brokstad KA, Jonsson MV, Delaleu N, Skarstein K. Current concepts on Sjögren’s syndrome - classification criteria and biomarkers. Eur J Oral Sci. 2018;126(1):37–48.
Christodoulou MI, Kapsogeorgou EK, Moutsopoulos HM. Characteristics of the minor salivary gland infiltrates in Sjögren’s syndrome. J Autoimmun. 2010;34(4):400–7.
Fox RI. Sjögren’s syndrome. Lancet. 2005;366(9482):321–31.
Hansen A, Lipsky PE, Dörner T. Immunopathogenesis of primary Sjögren’s syndrome: implications for disease management and therapy. Curr Opin Rheumatol. 2005;17(5):558–65.
Youinou P, Pers JO. Disturbance of cytokine networks in Sjögren’s syndrome. Arthritis Res Ther. 2011;13(4):227.
Gor DO, Rose NR, Greenspan NS. TH1-TH2: a procrustean paradigm. Nat Immunol. 2003;4(6):503–5.
d’Arbonneau F, Pers JO, Devauchelle V, Pennec Y, Saraux A, Youinou P. BAFF-induced changes in B cell antigen receptor-containing lipid rafts in Sjögren’s syndrome. Arthritis Rheum. 2006;54(1):115–26.
Negrini S, Emmi G, Greco M, Borro M, Sardanelli F, Murdaca G, et al. Sjögren’s syndrome: a systemic autoimmune disease. Clin Exp Med. 2022;22(1):9–25.
Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133(5):775–87.
Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. N Engl J Med. 2009;361(9):888–98.
Tian Y, Yang H, Liu N, Li Y, Chen J. Advances in Pathogenesis of Sjögren’s syndrome. J Immunol Res. 2021;2021:5928232.
Saraux A. The point on the ongoing B-cell depleting trials currently in progress over the world in primary Sjögren’s syndrome. Autoimmun Rev. 2010;9(9):609–14.
Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512–25.
Gui L, He X, Tang L, Yao J, Pi J. Obesity and head and neck cancer risk: a mendelian randomization study. BMC Med Genomics. 2023;16(1):200.
Lawlor DA, Harbord RM, Sterne JA, Timpson N, Davey Smith G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27(8):1133–63.
Burgess S, Dudbridge F, Thompson SG. Combining information on multiple instrumental variables in mendelian randomization: comparison of allele score and summarized data methods. Stat Med. 2016;35(11):1880–906.
Liu X, Tong X, Zou Y, Lin X, Zhao H, Tian L, et al. Mendelian randomization analyses support causal relationships between blood metabolites and the gut microbiome. Nat Genet. 2022;54(1):52–61.
Yeung CHC, Au Yeung SL, Schooling CM. Association of autoimmune diseases with Alzheimer’s disease: a mendelian randomization study. J Psychiatr Res. 2022;155:550–8.
Cao Y, Lu H, Xu W, Zhong M. Gut microbiota and Sjögren’s syndrome: a two-sample mendelian randomization study. Front Immunol. 2023;14:1187906.
Zhao M, Wei F, Li H, Wang Z, Wang S, Liu Y, et al. Serum vitamin D levels and Sjogren’s syndrome: bi-directional mendelian randomization analysis. Arthritis Res Ther. 2023;25(1):79.
Zhao JH, Stacey D, Eriksson N, Macdonald-Dunlop E, Hedman ÅK, Kalnapenkis A, et al. Genetics of circulating inflammatory proteins identifies drivers of immune-mediated disease risk and therapeutic targets. Nat Immunol. 2023;24(9):1540–51.
Roescher N, Tak PP, Illei GG. Cytokines in Sjögren’s syndrome. Oral Dis. 2009;15(8):519–26.
Felten R, Devauchelle-Pensec V, Seror R, Duffau P, Saadoun D, Hachulla E, et al. Interleukin 6 receptor inhibition in primary Sjögren syndrome: a multicentre double-blind randomised placebo-controlled trial. Ann Rheum Dis. 2021;80(3):329–38.
Liang Y, Zhang Z, Li J, Luo W, Jiang T, Yang Z. Association between IL-7 and primary Sjögren’s syndrome: a single-center study and a systematic scoping review. Int Immunopharmacol. 2022;108:108758.
Lin X, Wang X, Xiao F, Ma K, Liu L, Wang X, et al. IL-10-producing regulatory B cells restrain the T follicular helper cell response in primary Sjögren’s syndrome. Cell Mol Immunol. 2019;16(12):921–31.
Qi J, Li D, Shi G, Zhang X, Pan Y, Dou H, et al. Interleukin–12 exacerbates Sjögren’s syndrome through induction of myeloid–derived suppressor cells. Mol Med Rep. 2019;20(2):1131–8.
Sisto M, Lorusso L, Lisi S. Interleukin-15 as a potential new target in Sjögren’s syndrome-associated inflammation. Pathology. 2016;48(6):602–7.
Zhang LW, Zhou PR, Wei P, Cong X, Wu LL, Hua H. Expression of interleukin-17 in primary Sjögren’s syndrome and the correlation with disease severity: a systematic review and meta-analysis. Scand J Immunol. 2018;87(4):e12649.
Lavoie TN, Stewart CM, Berg KM, Li Y, Nguyen CQ. Expression of interleukin-22 in Sjögren’s syndrome: significant correlation with disease parameters. Scand J Immunol. 2011;74(4):377–82.
Soyfoo MS, Nicaise C. Pathophysiologic role of Interleukin-33/ST2 in Sjögren’s syndrome. Autoimmun Rev. 2021;20(3):102756.
Hernández-Molina G, Michel-Peregrina M, Hernández-Ramírez DF, Sánchez-Guerrero J, Llorente L. Chemokine saliva levels in patients with primary Sjögren’s syndrome, associated Sjögren’s syndrome, pre-clinical Sjögren’s syndrome and systemic autoimmune diseases. Rheumatology (Oxford). 2011;50(7):1288–92.
Hijazi Z, Wallentin L, Lindbäck J, Alexander JH, Connolly SJ, Eikelboom JW, et al. Screening of multiple biomarkers Associated with ischemic stroke in Atrial Fibrillation. J Am Heart Assoc. 2020;9(24):e018984.
Siegbahn A, Lindbäck J, Hijazi Z, Åberg M, Alexander JH, Eikelboom JW, et al. Multiplex protein screening of biomarkers associated with major bleeding in patients with atrial fibrillation treated with oral anticoagulation. J Thromb Haemost. 2021;19(11):2726–37.
Mbatchou J, Barnard L, Backman J, Marcketta A, Kosmicki JA, Ziyatdinov A, et al. Computationally efficient whole-genome regression for quantitative and binary traits. Nat Genet. 2021;53(7):1097–103.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75.
Choi KW, Chen CY, Stein MB, Klimentidis YC, Wang MJ, Koenen KC, et al. Assessment of Bidirectional relationships between physical activity and depression among adults: a 2-Sample mendelian randomization study. JAMA Psychiatry. 2019;76(4):399–408.
Yang J, Yan B, Zhao B, Fan Y, He X, Yang L, et al. Assessing the Causal effects of human serum metabolites on 5 Major Psychiatric disorders. Schizophr Bull. 2020;46(4):804–13.
Gill D, Brewer CF, Monori G, Trégouët DA, Franceschini N, Giambartolomei C, et al. Effects of genetically determined Iron status on risk of venous thromboembolism and carotid atherosclerotic disease: a mendelian randomization study. J Am Heart Assoc. 2019;8(15):e012994.
Burgess S, Thompson SG. Interpreting findings from mendelian randomization using the MR-Egger method. Eur J Epidemiol. 2017;32(5):377–89.
Hartwig FP, Davey Smith G, Bowden J. Robust inference in summary data mendelian randomization via the zero modal pleiotropy assumption. Int J Epidemiol. 2017;46(6):1985–98.
Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife. 2018;7(.
Lee EY, Lee ZH, Song YW. CXCL10 and autoimmune diseases. Autoimmun Rev. 2009;8(5):379–83.
Antonelli A, Ferrari SM, Giuggioli D, Ferrannini E, Ferri C, Fallahi P. Chemokine (C-X-C motif) ligand (CXCL)10 in autoimmune diseases. Autoimmun Rev. 2014;13(3):272–80.
Engel MA, Neurath MF. Anticancer properties of the IL-12 family–focus on colorectal cancer. Curr Med Chem. 2010;17(29):3303–8.
Antonelli A, Rotondi M, Fallahi P, Romagnani P, Ferrari SM, Paolicchi A, et al. Increase of interferon-gamma inducible alpha chemokine CXCL10 but not beta chemokine CCL2 serum levels in chronic autoimmune thyroiditis. Eur J Endocrinol. 2005;152(2):171–7.
Frigerio S, Junt T, Lu B, Gerard C, Zumsteg U, Holländer GA, et al. Beta cells are responsible for CXCR3-mediated T-cell infiltration in insulitis. Nat Med. 2002;8(12):1414–20.
Dauphinée MJ, Tovar Z, Ballester A, Talal N. The expression and function of CD3 and CD5 in patients with primary Sjögren’s syndrome. Arthritis Rheum. 1989;32(4):420–9.
Zumla A, Mathur M, Stewart J, Wilkinson L, Isenberg D. T cell receptor expression in Sjögren’s syndrome. Ann Rheum Dis. 1991;50(10):691–3.
Ichikawa Y, Shimizu H, Yoshida M, Takaya M, Arimori S. T cells bearing gamma/delta T cell receptor and their expression of activation antigen in peripheral blood from patients with Sjögren’s syndrome. Clin Exp Rheumatol. 1991;9(6):603–9.
Martín-Nares E, Hernández-Molina G, Lima G, Hernández-Ramírez DF, Chan-Campos I, Saavedra-González V, et al. Tear levels of IL-7, IL-1α, and IL-1β may differentiate between IgG4-related disease and Sjögren’s syndrome. Clin Rheumatol. 2023;42(4):1101–5.
Xu T, Xie W, Ma Y, Zhou S, Zhang L, Chen J, et al. Leptin/OB-R pathway promotes IL-4 secretion from B lymphocytes and induces salivary gland epithelial cell apoptosis in Sjögren’s syndrome. Oncotarget. 2017;8(38):63417–29.
Brayer JB, Cha S, Nagashima H, Yasunari U, Lindberg A, Diggs S, et al. IL-4-dependent effector phase in autoimmune exocrinopathy as defined by the NOD.IL-4-gene knockout mouse model of Sjögren’s syndrome. Scand J Immunol. 2001;54(1–2):133–40.
Saito K, Mori S, Date F, Ono M. Sjögren’s syndrome-like autoimmune sialadenitis in MRL-Faslpr mice is associated with expression of glucocorticoid-induced TNF receptor-related protein (GITR) ligand and 4-1BB ligand. Autoimmunity. 2013;46(4):231–7.
Zhou J, You BR, Yu Q. Agonist-induced 4-1BB activation prevents the development of Sjӧgren’s syndrome-like sialadenitis in non-obese diabetic mice. Biochim Biophys Acta Mol Basis Dis. 2020;1866(3):165605.
Cruz-Tapias P, Rojas-Villarraga A, Maier-Moore S, Anaya JM. HLA and Sjögren’s syndrome susceptibility. A meta-analysis of worldwide studies. Autoimmun Rev. 2012;11(4):281–7.
Brkic Z, Versnel MA. Type I IFN signature in primary Sjögren’s syndrome patients. Expert Rev Clin Immunol. 2014;10(4):457–67.
Fernandez-Ruiz R, Niewold TB. Type I interferons in autoimmunity. J Invest Dermatol. 2022;142(3 Pt B):793–803.
Furuzawa-Carballeda J, Sánchez-Guerrero J, Betanzos JL, Enriquez AB, Avila-Casado C, Llorente L, et al. Differential cytokine expression and regulatory cells in patients with primary and secondary Sjögren’s syndrome. Scand J Immunol. 2014;80(6):432–40.
Acknowledgements
We express our gratitude to all SCALLOP Consortium studies for providing open access to the summary association statistics data. Additionally, our heartfelt appreciation goes out to all the investigators and participants of the UK Biobank for making individual-level datasets publicly accessible and for their invaluable contributions to these studies.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
Wenbin Shi contributed to writing of this manuscript, formal analysis and visualization, conceptualization, supervision, methodology, project administration, review & editing of this manuscript. Yuli Xu and Anan Zhang contributed to resources and validation. Xiqun Jia contributed to methodology. Ziyang Hu contributed methodology, project administration, and review & editing of this manuscript. Shuhua Liu contributed to conceptualization, methodology, project administration, and review & editing. All authors gave final approval and agree to be accountable for all aspects of the work.
Corresponding authors
Ethics declarations
Ethical approval and consent to participants
All the data used in this research can be found in public databases. No additional ethical approval was required.
Consent for publication
The manuscript is approved by all authors for publication.
Competing interests
No competing interest exits in the submission of this manuscript.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
Cite this article
Shi, W., Xu, Y., Zhang, A. et al. Inflammatory cytokines and their potential role in Sjogren’s syndrome risk: insights from a mendelian randomization study. Adv Rheumatol 64, 14 (2024). https://doi.org/10.1186/s42358-024-00354-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42358-024-00354-2