
Abstract
Background
Mucopolysaccharidoses IVA is an autosomal recessive lysosomal storage disease resulting in skeletal and cartilage dysplasia. Hematopoietic stem cell transplantation is a good therapeutic option for MPS IV. Here we report the first application of PGD test for MPS IVA and HLA with the purpose of HSCT for the affected son in a family with consanguineous marriage. Haplotype analysis of linked STR markers in GALNS gene and HLA loci as well as variant detection by cycle sequencing were included in our PGD test.
Results
Two out of nine embryos were transferrable. The second embryo transfer was successful and resulted in the pregnancy of one healthy and HLA matched girl.
Conclusions
Preimplantation genetic diagnosis could be considered as a noninvasive clinical option for families with a mucopolysaccharidoses IVA patient to have a healthy child that is HLA-matched with the patient in need of hematopoietic stem cell transplantation. In lack of an appropriate hematopoietic stem cell donor the importance of preimplantation genetic diagnosis is much more significant too.
Similar content being viewed by others


Background
Mucopolysaccharidoses IVA (MPS IVA) or Morquio A syndrome is an autosomal recessive lysosomal storage disorder caused by mutations in GALNS gene on chromosome 16q24. This gene encodes N-acetylgalactosamine-6-sulfatase enzyme. In MPS IVA, lysosomal degradation of keratan sulphate and chondroitin-6-sulphate is impaired (Akyol et al. 2019). Its clinical feature includes short stature, skeletal and joint abnormalities including genu valgum, joint hypermobility, hip dislocation and dysplasia, dental anomalies, corneal clouding, hearing loss, pectus carinatum, spinal cord compression, spinal instability and thoracolumbar kyphoscoliosis. MPS IVA has variable severity, but patients with the severe phenotype usually die in their second or third decade of life. Respiratory impairment, cardiovascular disease and spinal cord instability are the main cause of morbidity and mortality (Akyol et al. 2019; Bertolin et al. 2021; Diaz-Ordoñez et al. 2022).
Hematopoietic stem cell transplantation (HSCT) and enzyme replacement therapy (ERT) are two main treatments for MPS which slow the progression of this disease and improve quality of life (Taylor et al. 2019).
In HSCT, the donor leukocytes go to the host tissues and supply the endogenous enzyme, while in the ERT (elosulfase alpha for MPS IVA) which is a lifelong therapy, exogenous enzymes are transported to host tissues. Although ERT can improve some clinical symptoms of the patients, it cannot pass through the blood–brain barrier and so cannot improve neurological implications. While in HSCT, donor-derived hematopoietic cells can pass through the blood–brain barrier and differentiate into microglia (Aldenhoven et al. 2008; Qu et al. 2022). Even if the ERT is started at an early stage, its effect is limited on skeletal dysplasia in MPS IVA patients. Although according to some studies HSCT may affect on bone, there is no evidence to improve bone growth if the HSCT starts after 4 years of age (Akyol et al. 2019; Sawamoto et al. 2020; Wang et al. 2016; Yabe et al. 2016). Also there are risks of graft versus host disease (GVHD) and rejection in HSCT and it needs a donor who is HLA matched to the patient (Sawamoto et al. 2020).
Preimplantation genetic diagnosis (PGD) makes it possible to select the healthy embryos before implantation in families with known disease causing mutations. PGD was applied for human more than 3 decades ago for the first time (Handyside et al. 1990; Verlinsky et al. 1990). Since then, PGD has been used for diagnosis of many genetic disorders such as hearing impairment, alpha and beta thalassaemia, Steroid-Resistant Nephrotic Syndrome, Neurofibromatosis, Neurodegenerative disorders, inherited heart diseases, etc., in the embryos (Chen et al. 2021; He et al. 2021; Khordadpoor Deilamani and Akbari 2019a, b; Merker et al. 2015; Sciorio et al. 2021; Yeates et al. 2022).
PGD has been also applied for HLA typing. The HLA (human leukocyte antigen) genes located on chromosome 6, encode antigen presenting proteins on the cell surface and have a main role in the immune system performance. HLA complex is responsible for GVHD and rejection after HSCT and organ or tissue transplantation (Alelign et al. 2018; Mori et al. 2021; Wiebe and Nickerson 2020). Due to high diversity of HLAs, the probability that two unrelated persons be HLA matched is very low and HLA-identical siblings provide the best opportunity to achieve a successful transplantation. When there is no HLA matched donor for a patient, PGD can be offered for the therapeutic purpose (Fernández et al. 2014; Kakourou et al. 2019).
Here we report the first use of PGD for both MPS IVA and HLA typing in a family with an affected son in order to select and transfer the unaffected embryos that are HLA matched with the patient too.
Methods
Patients
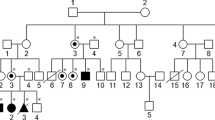
A couple (20-year-old female and 34-year-old male) who were distant relatives and having a 3-year-old son affected with MPS IVA were referred to the PGD center of Tehran Medical Genetics Laboratory. The homozygous mutation of c.319G>A (p.Ala107Thr) in exon 3 of GALNS gene had been found in the affected son by whole exome sequencing (WES) in the same laboratory. The family members were HLA typed with SSP (using Olerup SSP HLA Typing Kits) and/ or NGS method (Using AllType-NGS kit from One Lambda Inc. Thermofischer) and no one had matched HLA with the patient.
After PGD counseling, the informed written consent was obtained from the patient's parents. The local ethics committee approved this study.
In vitro fertilization
The In Vitro Fertilization procedure was performed at Avecina IVF center. Nine embryos were produced. Cells were biopsied from each embryo, washed in PBS and were transferred to our PGD center in 2.5 µl of PBS.
Whole genome amplification
Picoplex WGA kit (Rubicon Genomics) was used to perform whole genome amplification (WGA) on blastomeres as well as positive and negative control samples in 3 steps of cell lysis/DNA extraction, preamplification and amplification. Then QIAquick PCR purification kit from QIAGEN was used to purify the WGA products.
Cycle sequencing and haplotype analysis
Two different tests including Sanger sequencing of exon 3 of GALNS gene and linkage analysis by STR markers were used to assess the transmission of c.319G>A (p.Ala107Thr) mutation in GALNS gene to the embryos. Six dinucleotide short tandem repeats (STR) markers (rs58441312, rs1555521601, rs56406234, rs373372968, D16S3026 and D16S3121) encompassing the GALNS gene were included in linkage analysis.
Also a panel of 11 STR markers located inside the HLA locus was used to test the HLA haplotypes (Table 1). Forward primers of the STR markers were labeled with FAM or HEX fluorescent dyes.
The markers were amplified by several multiplex PCR and the fragment separation was done using Applied Biosystems 3130 Genetic Analyzer, Hi-Di formamide and GS 500 Liz size standard. The GeneMarker software was used to analyze the data.
Results
Sanger sequencing confirmed the c.319G>A (p.Ala107Thr) mutation which was detected by the whole exome sequencing in GALNS gene (Fig. 1). The segregation of the mutation was also confirmed in the family.

Homozygous mutation of c.319G>A (p.Ala107Thr) in GALNS gene in the Patient
HLA typing showed that the patient’s father was homozygous for HLA loci except for the HLA-DPB1 (DPB1*02:01:02/DPB1*04:01:01). Based on ImMunoGeneTics/HLA (IMGT/HLA) website (DPB1 T-Cell Epitope Algorithm v1.0 2012), these HLA-DPB1 alleles were permissive mismatch. So the embryos had the chance to inherit any paternal HLA-DPB1 allele for HSCT if they are matched with the donor for HLA-A, B, C, DRB1, and DQB1 loci (10/10) (Table 2).
In pre-PGD the haplotypes of the STR markers which were linked to the mutated GALNS allele as well as HLA loci were inferred by investigating the affected son, his parents and some other family members.
For PGD, the biopsied cells of nine embryos were received from the IVF center.
The WGA products were then used in order to determine the mutation status of the embryos. The embryos that had at least one normal allele in GALNS gene were tested with STR markers in HLA loci (Figs. 2 and 3). The summarized results are presented as follows (Table 3).

Haplotype analysis for STR markers flanking the GALNS gene and sequencing results of GALNS mutation

Haplotype analysis for STR markers in the HLA loci
Embryos 3 and 7 were recommended to be transferred. The first transfer was unsuccessful. The conception was successful at the second transfer. Prenatal diagnosis was done at 12th week of gestation and confirmed the conception of a girl who was heterozygous for normal allele and has matched-HLA with the affected son. The fetus is not born yet. After its birth HSCT will be performed from the Umbilical cord blood.
Discussion
MPS IVA is a lysosomal storage disease which can be fatal in patients with severe phenotypes (Sawamoto et al. 2020; Wang et al. 2016; Yabe et al. 2016).
The first successful case report of HSCT for MPS IVA was reported in 2014 (Chinen et al. 2014).
In 2020, HSCT was suggested as a useful treatment option for MPS IVA patients that could improve pulmonary performance, density of bone mineral and daily life activities and could reduce the need to surgeries in these patients. However HSCT has the risk of mortality and morbidity more than ERT (Sawamoto et al. 2020).
In 2022 Qu et al. reported that the lysosomal enzyme restored to the normal level after HSCT in patients with MPS IVA and VI. Also the functions of respiratory and nervous systems were improved (Qu et al. 2022).
PGD for Morquio syndrome was done previously in order to prevent the birth of new affected child (Qubbaj et al. 2008). However, there is no report of PGD with the purpose of treatment for this disorder. Here we report the first application of PGD for MPS IVA and HLA typing for future HSCT treatment for the affected child in a family. SSP and NGS-based HLA typing revealed there is no appropriate donor in this family. In the HLA registry there were some HLA matched donor oversees. However the patient's parents could not accept the risk of any morbidity or mortality of HSCT from a non-relative donor to their son and requested for the PGD test.
The ESHRE guideline recommended using two upstream and two downstream markers that are linked to the disease causing gene in indirect mutation analysis to reduce the risk of Allele dropout (ADO) and no diagnosis results in PGD. Also for HLA typing at least one STR marker located upstream of HLA-A, one between HLA-A and HLA-B, one between HLA-B and HLA-DRA, one between HLA-DRA and HLA-DQB1 and one downstream of HLA-DQB1 should be used to be able to assess the recombination in the HLA region. Applying two markers makes the test more powerful (Harton et al. 2011).
We used 3 upstream and 3 downstream STR markers flanking the GALNS gene and totally eleven markers between HLA loci. Allele dropout (ADO) and Locus dropout (LDO) were observed in different loci (Fig. 2 and 3). Also, due to incomplete adenylation in PCR, analysis of rs58441312 marker which is an AT repeat was difficult in most samples especially in WGA products. However, using sufficient number of STR markers in combination with Sanger sequencing made it possible to determine the genotypes of embryos.
Embryos 3, 6, 7 and 8 had at least one normal allele. These embryos as well as embryo 4 for which the zygosity of the mutation was not determined in the first try, were tested for STR markers in HLA loci which resulted in determining two HLA matched embryos (embryos 3 and 7).
The second transfer of the embryos resulted in pregnancy. Prenatal diagnosis in 12th week of gestation confirmed the conception of a heterozygous and HLA matched female which was also normal for the copy number of chromosomes 13, 18, 21, and X based on the QF-PCR Method.
As far as moral issues regarding PGD for HSCT is concerned, it is argued that since performing HSCT from an existing child to cure a sibling is acceptable, so PGD for HSCT is moral too (Pennings et al. 2002). Also when deciding to have children for less important reasons such as improving the marriage of disconnected couples is acceptable, wishing to conceive a child with PGD for HLA typing to treat another child is not morally wrong (Nickel and Kamani 2018). When there is no proper treatment except HSCT, the HSCT has high success rate, there is no HLA matched donor or HSCT from HLA matched sibling has advantage in comparison with nonrelative donors and HSCT can wait at least 9–12 months for the birth of the donor child, the conditions are appropriate for PGD for HLA typing (Pennings et al. 2002).
This report of PGD for MPS IVA and HLA may accelerate the setup of this test in other laboratories especially in countries with high frequency of consanguineous marriages and higher risk of genetic disorders such as MPS IVA.
Conclusions
In conclusion, despite the complexity of the procedure, PGD for MPS IVA and HLA typing can be considered as an option for curing the MPS IVA patients with HSCT. This should be considered especially for cases with no HLA matched donor.
Availability of data and material
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- HSCT:
-
Hematopoietic stem cell transplantation
- ERT:
-
Enzyme replacement therapy
- MPS IVA:
-
Mucopolysaccharidoses IVA
- GVHD:
-
Graft versus host disease
- PGD:
-
Preimplantation genetic diagnosis
- HLA:
-
Human leukocyte antigen
- WES:
-
Whole exome sequencing
- WGA:
-
Whole genome amplification
- STR:
-
Short tandem repeats
- ADO:
-
Allele dropout
- LDO:
-
Locus dropout
References
Akyol MU, Alden TD, Amartino H, Ashworth J, Belani K, Berger KI, Borgo A, Braunlin E, Eto Y, Gold JI (2019) Recommendations for the management of MPS IVA: systematic evidence-and consensus-based guidance. Orphanet J Rare Dis 14(1):1–25
Aldenhoven M, Boelens J, de Koning TJ (2008) The clinical outcome of Hurler syndrome after stem cell transplantation. Biol Blood Marrow Transplant 14(5):485–498
Alelign T, Ahmed MM, Bobosha K, Tadesse Y, Howe R, Petros B (2018) Kidney transplantation: the challenge of human leukocyte antigen and its therapeutic strategies. J Immunol Res. https://doi.org/10.1155/2018/5986740
Bertolin J, Sánchez V, Ribera A, Jaén ML, Garcia M, Pujol A, Sánchez X, Muñoz S, Marcó S, Pérez J (2021) Treatment of skeletal and non-skeletal alterations of Mucopolysaccharidosis type IVA by AAV-mediated gene therapy. Nat Commun 12(1):1–14
Chen H-L, Lin P-H, Chiang Y-T, Huang W-J, Lin C-F, Ma G-C, Chang S-P, Fan J-Y, Lin S-Y, Wu C-C (2021) Preimplantation genetic diagnosis in hereditary hearing impairment. Diagnostics 11(12):2395
Chinen Y, Higa T, Tomatsu S, Suzuki Y, Orii T, Hyakuna N (2014) Long-term therapeutic efficacy of allogenic bone marrow transplantation in a patient with mucopolysaccharidosis IVA. Mol Genet Metab Rep 1:31–41
Diaz-Ordoñez L, Candelo E, Silva-Cuero K, Saldarriaga W, Murgašová L, Magner M, Pachajoa H (2022) Hearing loss in patients with morquio syndrome: protocol for a scoping review. JMIR Research Protocols 11(6):e32986
DPB1 T-Cell Epitope Algorithm v1.0. https://www.ebi.ac.uk/ipd/imgt/hla/matching/dpb_v1/. Accessed April 2012
Fernández RM, Peciña A, Lozano-Arana MD, Sánchez B, Guardiola J, García-Lozano JC, Borrego S, Antiñolo G (2014) Experience of preimplantation genetic diagnosis with HLA matching at the University Hospital Virgen del Rocío in Spain: technical and clinical overview. Biomed Res Int. https://doi.org/10.1155/2014/560160
Handyside AH, Kontogianni EH, Hardy K, Winston RM (1990) Pregnancies from biopsied human preimplantation embryos sexed by Y-specific DNA amplification. Nature 344(6268):768–770
Harton G, De Rycke M, Fiorentino F, Moutou C, SenGupta S, Traeger-Synodinos J, Harper J (2011) ESHRE PGD consortium best practice guidelines for amplification-based PGD. Hum Reprod 26(1):33–40
He T-W, Lu J, Chen C-Q, Zhou W-N, Li J-S, Dong Y-Q, Yin A (2021) Preimplantation genetic diagnosis of α/β complex thalassemia by next generation sequencing. Zhongguo Shi Yan Xue Ye Xue Za Zhi 29(4):1275–1279
Kakourou G, Mamas T, Vrettou C, Traeger-Synodinos J (2019) Preimplantation genetic testing for HLA-matching: an overview of clinical application and utility. OBM Genet 3(3):1–1
Khordadpoor Deilamani F, Akbari MT (2019) First report of preimplantation genetic diagnosis for steroid-resistant nephrotic syndrome. J Human Gen Genom. https://doi.org/10.5812/jhgg.109109
Khordadpoor Deilamani F, Akbari MT (2019) Preimplantation genetic diagnosis for beta Thalassemia. J Human Gen Genom. https://doi.org/10.5812/jhgg.109503
Merker VL, Murphy TP, Hughes JB, Muzikansky A, Hughes MR, Souter I, Plotkin SR (2015) Outcomes of preimplantation genetic diagnosis in neurofibromatosis type 1. Fertil Steril 103(3):761-768.e761
Mori A, Murata S, Tashiro N, Tadokoro T, Okamoto S, Otsuka R, Wada H, Murata T, Takahashi T, Seino K-i (2021) Establishment of human leukocyte antigen-mismatched immune responses after transplantation of human liver bud in humanized mouse models. Cells 10(2):476
Nickel RS, Kamani NR (2018) Ethical challenges in hematopoietic cell transplantation for sickle cell disease. Biol Blood Marrow Transplant 24(2):219–227
Pennings G, Schots R, Liebaers I (2002) Ethical considerations on preimplantation genetic diagnosis for HLA typing to match a future child as a donor of haematopoietic stem cells to a sibling. Hum Reprod 17(3):534–538
Qu Y, Liu H, Wei L, Nie S, Ding W, Liu S, Liu H, Jiang H (2022) The outcome of allogeneic hematopoietic stem cell transplantation from different donors in recipients with mucopolysaccharidosis. Front Pediatr. https://doi.org/10.3389/fped.2022.877735
Qubbaj W, Al-Aqeel AI, Al-Hassnan Z, Al-Duraihim A, Awartani K, Al-Rejjal R, Coskun S (2008) Preimplantation genetic diagnosis of Morquio disease. Prenat Diagn 28(10):900–903
Sawamoto K, Álvarez González JV, Piechnik M, Otero FJ, Couce ML, Suzuki Y, Tomatsu S (2020) Mucopolysaccharidosis IVA: diagnosis, treatment, and management. Int J Mol Sci 21(4):1517
Sciorio R, Aiello R, Irollo AM (2021) Preimplantation genetic diagnosis (PGD) as a reproductive option in patients with neurodegenerative disorders. Reprod Biol 21(1):100468
Taylor M, Khan S, Stapleton M, Wang J, Chen J, Wynn R, Yabe H, Chinen Y, Boelens JJ, Mason RW (2019) Hematopoietic stem cell transplantation for mucopolysaccharidoses: past, present, and future. Biol Blood Marrow Transplant 25(7):e226–e246
Verlinsky Y, Ginsberg N, Lifchez A, Valle J, Moise J, Strom CM (1990) Analysis of the first polar body: preconception genetic diagnosis. Hum Reprod 5(7):826–829
Wang J, Luan Z, Jiang H, Fang J, Qin M, Lee V, Chen J (2016) Allogeneic hematopoietic stem cell transplantation in thirty-four pediatric cases of mucopolysaccharidosis: a 10-year report from the China children transplant group. Biol Blood Marrow Transplant 22(11):2104–2108
Wiebe C, Nickerson PW (2020) Human leukocyte antigen molecular mismatch to risk stratify kidney transplant recipients. Curr Opin Organ Transplant 25(1):8–14
Yabe H, Tanaka A, Chinen Y, Kato S, Sawamoto K, Yasuda E, Shintaku H, Suzuki Y, Orii T, Tomatsu S (2016) Hematopoietic stem cell transplantation for Morquio A syndrome. Mol Genet Metab 117(2):84–94
Yeates L, McDonald K, Burns C, Semsarian C, Carter S, Ingles J (2022) Decision-making and experiences of preimplantation genetic diagnosis in inherited heart diseases: a qualitative study. Eur J Hum Genet 30(2):187–193
Acknowledgements
We thank the personnel of Tehran Medical Genetics Laboratory for their support.
Funding
This project was financially supported by Tehran Medical Genetics Laboratory.
Author information
Authors and Affiliations
Contributions
FK designed and performed the test, analyzed and interpreted the data and wrote the manuscript. MTA provided scientific guidance and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The informed consent was obtained from the couple and the study was approved by the local ethics committee.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Khordadpoor Deilamani, F., Akbari, M.T. First report of preimplantation genetic diagnosis of mucopolysaccharidoses IVA and HLA typing for hematopoietic stem cell transplantation. Bull Natl Res Cent 46, 283 (2022). https://doi.org/10.1186/s42269-022-00972-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42269-022-00972-0