
Abstract
Background
Maize is one of the important cereal food crops in the world. High temperature stress causes adverse influence on plant growth. When plants are exposed to high temperatures, they produce heat shock proteins (HSPs), which may impart a generalized role in tolerance to heat stress. Proteome analysis was performed in plant to assess the changes in protein types and their expression levels under abiotic stress. The purpose of the study is to explore which proteins are involved in the response of the maize plant to heat shock treatment.
Results
We investigated the responses of abundant proteins of maize leaves, in an Egyptian inbred line of maize “K1”, upon heat stress through two-dimensional electrophoresis (2-DE) on samples of maize leaf proteome. 2-DE technique was used to recognize heat-responsive protein spots using Coomassie Brilliant Blue (CBB) and silver staining. In 2-D analysis of proteins from plants treated at 45 °C for 2 h, the results manifested 59 protein spots (4.3%) which were reproducibly detected as new spots where did not present in the control. In 2D for treated plants for 4 h, 104 protein spots (7.7%) were expressed only under heat stress. Quantification of spot intensities derived from heat treatment showed that twenty protein spots revealed clear differences between the control and the two heat treatments. Nine spots appeared with more intensity after heat treatments than the control, while four spots appeared only after heat treatments. Five spots were clearly induced after heat treatment either at 2 h or 4 h and were chosen for more analysis by LC-MSMS. They were identified as ATPase beta subunit, HSP26, HSP16.9, and unknown HSP/Chaperonin.
Conclusion
The results revealed that the expressive level of the four heat shock proteins that were detected in this study plays important roles to avoid heat stress in maize plants.
Similar content being viewed by others


Background
The most critical function of plant cell is to respond against stress induced for self-defense. This defense is brought about by modification in the pattern of gene expression: qualitative and quantitative changes in proteins leading to modulation of certain metabolic and defensive pathways. Recent proteome analysis performed in plant has provided new dimensions to evaluate the changes in protein types and their expression levels under abiotic stress. Specific and novel proteins have been identified, which play a role in heat shock. Beside specific protein production, plants respond to a variety of stresses by producing heat shock proteins (HSPs), indicating the plant’s adaptive mechanisms which HSPs use to protect cells against many stresses (Timperio et al. 2008). Cereal grain proteomics can provide valuable information regarding plant growth, nutritional status, adaptation to environmental stresses, and the role that grain proteins play in health disorders, such as coeliac disease (Bose et al. 2019).
Abril et al. (2011) found that proteomics provide a simultaneous and high throughput study of protein profiles in cells, tissues, or a living organism at any developmental state or under a specific environmental condition. Magdeldin et al. (2014) remarked that the gel-based proteomics is one of the most versatile methods for fractionating protein complexes. Along with these methods, 2-DE is commonly used to simultaneously fractionate, identify, and quantify proteins when coupled with MS identification. Westermeier (2014) reported that separating proteins according to the combination of isoelectric focusing under denaturing conditions and sodium dodesil sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) delivers the highest resolution of all bio-analytic techniques. Tan et al. (2017) reported that proteomics allows us to expansively analyze crop proteins and facilitate to understand the functions of the genes. MS can now identify a wide range of proteins and monitor their modulation during plant growth and development, as well as during responses to abiotic and biotic stresses.
HSPs are molecular chaperones that regulate the folding, localization, accumulation, and degradation of protein molecules in the plant species, which they play a wide role in many cellular processes, which may impart a generalized role in tolerance to various environmental stresses (Feder and Hofmann 1999). According to their approximate size, HSPs are grouped into HSP100, HSP90, HSP70, HSP60, and sHSPs (Sanmiya et al. 2004). The transcription of genes encoding HSPs is controlled by regulatory proteins called heat stress transcription factors (HSFs) located in the cytoplasm as inactive bodies (Hu et al. 2009). Rizhsky et al. (2004) studied response of plant to heat stress by analysis of Arabidopsis transcriptome, in which some genes appeared upregulated. The products of these genes included HSP100/ClpB, HSP90/HtpG, HSP70/DnaK, HSP60/GroEL, and small HSPs, confirming proteomic studies. These HSPs are proposed to act as molecular chaperones in protein quality control. Lee et al. (2007) investigated the responses of abundant and low-abundant proteins of rice leaves upon heat stress, performed 2-DE on PEG-fractionated supernatant and pellet samples of rice leaf proteome. A total of 48 differentially expressed proteins in samples taken after 12 or 24 h of heat exposure compared to controls were revealed, 18 of which were HSP (HSP70, Dnak-type molecular chaperone BiP, HSP100, Cpn60 and sHSPs).
The expression of HSP90 is developmentally regulated and responds to heat, cold, and salt stress in the tobacco plants (Krishna and Gloor 2001). Members of Hsp70 chaperones in Arabidopsis are expressed in response to stress such as heat, cold, and drought (Sung et al. 2001). Recent findings showed that the sHSP18.1 isolated from Pisum sativum, as well as HSP16.6 from Synechocystis sp. PCC6803 under in vitro conditions, binds to unfolded proteins and allows more refolding by HSP70/HSP100 complexes (Mogk et al. 2003). Chen et al. (2011) used 2-DE to identify combined drought- and heat-responsive protein spots in maize leaves. After Coomassie Brilliant Blue (CBB) staining, about 450 protein spots were reproducibly observed on each gel, wherein seven spots were expressed only under heat and combined drought and heat stress. Using MALDI-TOF mass spectrometry, a total of seven proteins were identified, including cytochrome b6-f complex iron sulfur subunit, sHSP17.4, sHSP17.2, sHSP26, guanine nucleotide-binding protein b-subunit-like protein, putative uncharacterized protein, and granule-bound starch synthase IIa. Moreover, the gene expression of three sHSPs was analyzed at the transcriptional level and showed that the three sHSPs were expressed under several treatments although their expression levels were obviously more enhanced by heat than by control.
Chen et al. (2014) studied HSP26.7, HSP23.2, HSP17.9, HSP17.4, and HSP16.9 in rice (Oryza sativa L.), which were upregulated in “Nipponbare” cultivar during seedling and anthesis stages in response to heat stress. The expressing levels of these five sHSPs in the heat-tolerant rice cultivar “Co39” were all significantly higher than that in the heat-susceptible rice cultivar “Azucena”. This indicated that the expressive level of these five sHSPs was positively related to the ability of rice plants to avoid heat stress. Thus, the expression level of these five sHSPs can be considered as biomarkers for screening rice cultivars to avoid heat stress. Klein et al. (2014) studied the expression and purification of two Class II sHSPs from Zea mays L. (cv. Oh43). The two proteins ZmHSP17.0-CII and ZmHSP17.8-CII oligomerize to form dodecamers at temperatures lower than heat shock (HS). There are significant differences between the two sHSPs during heat shock at 43 °C: ZmHSP17.8-CII dissociates into smaller oligomers than ZmHSP17.0-CII and is a more efficient chaperone with target protein citrate synthase, while ZmHSP17.0-CII is expressed during development. Hu et al. (2015) demonstrated the relevance of sHSP26 in protecting maize chloroplasts under heat stress and potentially involved in maize heat tolerance. 2-DE based proteomics, RNA interference (RNAi), and co-immunoprecipitation (Co-IP) were used to reveal chloroplast proteins interacting with sHSP26 and how sHSP26 functions under heat stress. After the silencing of sHSP26, a total of 45 protein spots from isolated protoplasts were changed in abundance, of which 33 spots are chloroplastic. Co-IP revealed that nine proteins possibly associated with sHSP26. In particular, four proteins, including ATP synthase subunit β, chlorophyll a–b binding protein, oxygen-evolving enhancer protein 1, and photosystem I reaction center subunit IV, strongly interacted with sHSP26 and greatly abundant under heat stress. The aim of this investigation is to explore which proteins are involved in the response of the maize plant to heat shock treatment and using them as molecular markers to improve heat stress tolerance in the maize inbred lines.
Materials and methods
An Egyptian inbred line of maize (Zea mays L.) “K1” was used in this study which was developed by the plant breeding group, Genetic and Cytology Department, National Research Centre, Giza, Egypt. The inbred “K1” is one of four Egyptian maize inbred lines (Zea mays L.) that were studied to identify heat shock proteins under heat stress at 45 °C as protein markers for detecting the thermo-tolerance inbred lines, which showed four bands of heat shock proteins with molecular weights of 82, 22, 17, and 10 kDa after exposing to 45 °C for 2 and 4 h which may be an indication of thermo-tolerance (Mahmoud et al. 2018).
Two-dimensional electrophoresis and analysis for “K1” line
Protein extraction
-
1.
The inbred line “K1” was grown for 2 weeks.
-
2.
The seedlings were exposed to HS (45 °C) for 2 and 4 h besides control (25 °C).
-
3.
Seedlings (0.2 g) were ground in liquid nitrogen and the powder added to 2 ml of lyses buffer containing 7 M urea, 2 M thiourea, 4% CHAPS, 18 mM Tris-HCl pH 8.0, and 4% Triton X-100 in presence of 53 u/mL DNase I, 4.9 u/mL RNase, and a cocktail of proteases inhibitors (1 mM PMSF, 0.1 mM pepstatin, 2 mM leupeptin, 1 mM E-64 and 1 mM aprotinin) were added to the solution. They were incubated at 4 °C for 20 min.
-
4.
Dithiothreitol (DTT) 14 mM was added and the samples were centrifuged for 20 min at 10000×g at 4 °C.
-
5.
Step 4 was repeated twice and protein content was quantified by assay of Bradford (1976) using the Bio-Rad protein assay with BSA as standard and equal loading amounts were confirmed on 1-D gels stained with CBB-R250.
The first-dimensional gel
-
1.
The samples containing 1 mg of proteins were diluted in rehydration solution (8 M urea, 18 mM 156 Tris-HCl pH 8.0, 4% w/v CHAPS, 0.5% v/v immobilized pH gradient (IPG) buffer (pH 4-7), 1.6% v/v DeStreak Reagent (GE Healthcare), and 0.002% w/v Bromophenol Blue) in a final volume of 350 μl and loaded onto pH 4–7, 18 cm IPG strips (DryStrips, GE Healthcare) for the first dimension.
-
2.
Isoelectric focusing (IEF) was performed at 500 V in gradient for 1 h 30 min, using EttanTMIPGphorTM IEF system (GE Healthcare).
-
3.
IPG strips were equilibrated with 50 mM Tris-HCl (pH 8.8), 6 M urea, 30% (v/v) glycerol, 2% SDS, a trace of Bromophenol Blue, and 10 mg/ml DTT during 15 min, followed by a second equilibration step with the same buffer containing 25 mg/ml iodoacetamide instead of DTT, for 15 min with shaking.
The second dimensional gel
-
1.
The strips were loaded and run on SDS-PAGE 13% poly acrylamide during 3–4 h at 100 V.
-
2.
Three experimental replicates were performed for each treatment and validated.
-
3.
Proteins were visualized with CBB-R250 staining (Bio-Rad) and by silver stain according to Shevchenko et al. (1996).
-
4.
The stained gels were scanned at a 300-dpi resolution with an image scanner desktop instrument and the Lab Scan application (GE Healthcare).
-
5.
Images were saved as Tag Image File Format (TIFF) and the spot detection and gel comparison were made with the Image Master Platinum version 5.1 Software (Amersham, Biosciences).
Protein identification by MS and database search
In-gel digestion
- 1.
Protein spots differentially expressed between the samples were manually excised from 2-D gels and digested with trypsin for further identification as described by Irar et al. (2010).
- 2.
Excised gel spots were washed sequentially with 50 mM ammonium bicarbonate (NH4HCO3) buffer and acetonitrile (ACN). The proteins were reduced and alkylated respectively, by treatment with 10 mM DTT in solution during 30 min at 56 °C, and another treatment with 55 mM of iodine acetamide.
- 3.
After sequential washings with NH4HCO3 buffer and ACN, proteins were digested overnight at 37 °C with 6 pmol of trypsin. Tryptic peptides were extracted from the gel matrix with 10% formic acid and ACN, the extracts were pooled and vacuum dried.
Acquisition of MS/MS spectra by LC-ESI-MSMS
The proteins excised from the 2D gels were either analyzed in a MALDI (matrix-assisted laser desorption/ionization)-TOF/TOF (4700 Proteomics Analyzer, Applied Biosystems) or LC-ESI (electrospray ionization)-QTOF (Q-TOF Global) mass photometers. For MALDI-TOF/MS analysis, the digested peptides were re-dissolved in 5 μl of 0.1% trifluoroacetic acid trifluoroacetic acid (TFA) in 50% ACN. Typically, 0.5 to 1 μl aliquot was mixed well with the same volume of a matrix solution, 2 to 5 mg/mL α-ciano-4-217 hydroxycinnamic acid in 0.1% TFA in 50% ACN and spotted to the MALDI plate. MS spectra were acquired in positive reflector mode (voltage of 20 KV in the source 1 and laser intensity ranged from 4800 to 5200). Five hundred shots by spectrum were accumulated.
Three major peaks were selected to be further characterized by MS/MS analysis. MS/MS spectra were acquired using collision-induced dissociation (CID) with atmospheric air as the collision gas. MS-MS 1 KV positive mode was used. MS and MS/MS spectrum from the same spot were merged in a single mgf file prior to submission for database searching. For LC-MS/MS analysis, online LC tandem mass spectrometry (Cap-LC-nano-225 ESI-Q-TOF) (Cap-LC, Micro-mass-Waters), the digested peptides samples were re-suspended in 25 μl of 1% formic acid solution and 4 μl were injected to chromatographic separation in reverse-phase capillary C18 column. The eluted peptides were ionized via coated nano-ES needles. A capillary voltage of 1800 to 2800 V was applied together with a cone voltage of 80 V. The collision in the CID was 20–35 eV and argon was employed as a collision gas.
Data analysis
Data were generated in PKL file format. PKL and mgf text files were submitted for database searching in a MASCOT search engine against non-redundant NCBI and SWISS-PROT Bank Databases.
Results
Two-dimensional electrophoresis analyses in the line “K1” leaves under heat stress
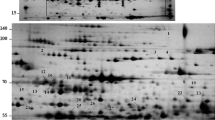
The proteomic analysis was carried out on extracted proteins from leaves of 2 weeks old treated at 45 °C for 4 h. The proteins were separated in range of pI 4–7 and molecular weights 14–66 kDa as shown in Fig. 1.

2-DE gel images of leaves protein extracted from K1 line treated for 4 h at 45 °C. An equal amount from total proteins (1 mg) was loaded in each gel, protein spots were detected with CBB-R250 and silver stain
The spots of proteins that were produced by 2-DE were detected after CBB-R250 and silver staining, in which the percentage of merge between the three gel replicates of each treatment beside the untreated plants was validated (higher than 60%). The proteome of sensitive vs. tolerant provenances showed a percentage of spot matching ranged from 67 to 76%. In addition, we checked individual spots with fold change ratios > 1.5 (P < 0.05) between different proteomes (Fig. 2). The three replicates gave a good percentage for all treatments and the control.

The correlation between the three gel replicates of each treatment. The merge between the three replicates of untreated plants (control) was 73%, for the treated plants at 45 °C for 2 h was 67%, while 76% was shown at 45 °C for 4 h
New proteins were induced in maize leaves of heat treatment plants as revealed in Fig. 3. In 2-DE analysis of proteins from plants treated at 45 °C for 2 h, the results manifested that 63% of spots merged with the control while 59 spots (4.3%) were produced as new spots where absent in the control. The analysis of 2-DE for treated plants for 4 h, showed that 60% of protein spots merged with control while 104 new spots (7.7%) were produced only in the treated plants. It is noted that there is a merge of 70% of proteins were induced upon heat treatment either for 2 h or 4 h, which 1.2% new spots appeared after treatment at 45 °C for 2 h and 3.2% new spots appeared after treatment at 45 °C for 4 h.

Venn diagrams show the mean number of matching spots between different sets of contrasting line after gel comparisons using the image master platinum software
Quantification of spot intensities derived from HS treatments in maize inbred “K1” is shown in Fig. 4. Twenty spots showed clear differences between the control and the two heat treatments. Nine spots numbers 1422, 1402, 1395, 1382, 982, 901, 829, 770, and 712 appeared more intensity after heat treatments than the control. Six spot numbers 1198, 1179, 862, 2777, 2782, and 2794 revealed more intensity in the control than in the heat treatments, while four spots appeared only after heat treatments which were undetected in the control. Five spots were clearly induced after heat treatment either at 2 h or 4 h and were chosen for more analysis (1402, 1395, 1090, 1080 and 982) which in 1090 and 1080 spots, the proteins are absent in the control (Fig. 5).

Graphic represents an intensity comparison of the representative samples of spots detected in 2-D gels of untreated K1 plants (blue), treated at 45 °C for 2 h (red) and 4 h (green). Grey arrows indicate spots that are isolated for protein identification

Detail of the differential intensities of chosen spots in response to heat treatment (a). Spots were chosen for LC-MSMS analysis (b)
The five spots picked off from the gel to identify by mass spectrometer (LC-MSMS) were treated by trypsin for digestion; then, the peptide mixtures were analyzed by liquid chromatography in capillary column and the peptides were exposed to mass analysis by LC-MSMS as shown in Fig. 6.

Protein identification by LC-MSMS procedure for the five picked off spots
The five proteins that identified as heat shock proteins by 2-DE and LC-MSMS were compared with the SWISS-PROT Bank Databases of National Center of Biological Information (NCBI). The results of comparison are shown in Table 1.
Features of the identified heat-induced proteins via SWISS-PROT Bank Databases of NCBI are as follows:
- 1.
ATPase beta subunit: This protein is present in chloroplasts and mitochondria which contain chaperonine-like domains.
- 2.
Unknown HSP/Chaperonin: Two isoforms of unknown proteins contain two regions of chaperonin-like that cooperate with other chaperones and ATPases to assist folding and assembly of proteins in the matrix of mitochondria and chloroplasts.
- 3.
HSP 26: Maize HSP26 is a nuclear-encoded and plastid-localized protein in maize as reported by Nieto-Sotelo et al. (1990). It has been described as an early induced heat-shock protein.
- 4.
HSP 16.9: Maize HSP16.9 is a cytosolic class I of sHSP.
Discussion
HSPs perform chaperone functions by stabilizing new proteins to ensure correct folding or by helping to refold proteins that were damaged by cell stress. This increase in expression is transcriptionally regulated. The transcription of genes encoding HSPs is controlled by regulatory proteins called HSFs (Hu et al. 2009). 2-D technique is used to separate ampholytic components, improving protein recognition, and to analyze and compare synthesis, turnover, and modification of many proteins during development or in response to environmental changes (Pergande and Cologna 2017, Schuster and Davies 1983). Chen et al. (2014) used 2-DE to identify combined drought- and heat-responsive protein spots in maize leaves, which found about 450 spots were reproducibly detected on each gel. They identified seven proteins by using MALDI-TOF mass spectrometry, including cytochrome b6-f complex iron sulfur subunit, sHSP17.4, sHSP17.2, and sHSP26. Klein et al. (2014) reported that sHSP17 and sHSP17.8 are molecular chaperones that protect maize cells from the effect of heat and other stresses. These results are in agreement with our results in which we used 2-DE technique and MALDI-TOF mass spectrometry to recognize and isolate the proteins involved in the response of the maize plant to HS treatment such as ATPase beta subunit, HSP/Chaperonin, HSP 26, and HSP 16.9.
It has been reported that mitochondrial and chloroplast ATPase are HS proteins (Chen et al. 2011). Recent transcriptoma studies demonstrate that ATPase, a complex of several proteins is heat-inducible in grasses (Li et al. 2013). HSP26 is a temperature-regulated molecular chaperone, where it forms large oligomeric complexes that are functionally activated by dissociation upon HS. It has been shown that proteins bound to HSP26 can be refolded to the native state in cooperation with HSP70 in an ATP-dependent reaction (Haslbeck et al. 1999, Ehrnsperger et al. 1997). Hu et al. (2015) demonstrated that chloroplast sHSP26 is abundant in maize leaves under HS and potentially involved in maize heat tolerance. They found that sHSP26 improved maize chloroplast performance under heat stress by interacting with specific proteins such as ATP synthase subunit β. Sun et al. (2012) reported that ZmHSP16.9 is expressed in root, leaf, and stem tissues under 40 °C treatment which is upregulated by HS and exogenous H2O2. Over expression of ZmHSP16.9 in transgenic tobacco conferred tolerance to heat and oxidative stresses, this supports the positive role of ZmHSP16.9 in its response to heat stress in plant. Chen et al. (2014) found, in rice, that HSPs including HSP16.9 were upregulated seedling stage in response to HS. These results coincided with our results that emphasized the role of ATPase beta subunit, HSP26, and HSP16.9 in maize heat tolerance.
Conclusions
Proteome analysis performed in plant has determined the changes in protein types and their expression levels under abiotic stress. The responses of abundant proteins of maize leaves to heat stress were investigated through two-dimensional electrophoresis (2-DE). In 2-D analysis of proteins from plants treated at 45 °C for 2 h and 4 h, the results manifested 59 protein spots (4.3%) and 104 protein spots (7.7%) were reproducibly detected as new spots were not present in the control. Quantification of spot intensities showed that twenty protein spots revealed clear differences between the control and the two heat treatments. Nine spots appeared more intensity after heat treatments than the control, while four spots appeared only after heat treatments. Five spots were clearly induced after heat treatment either at 2 h or 4 h and were chosen to more analysis by LC-MSMS. They were identified as ATPase beta subunit, HSP26, HSP16.9, and unknown HSP/Chaperonin. This revealed that the expressive level of these four proteins play important roles to avoid heat stress in maize plants. Moreover, the expression of HSPs can be considered as biomarkers for screening maize varieties to heat stress.
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- 2-DE:
-
Two-dimension electrophoresis
- BSA:
-
Bovine serum albumin
- ACN:
-
Acetonitrile
- CBB:
-
Coomassie Brilliant Blue
- CID:
-
Collision-induced dissociation
- Co-IP:
-
Co-immunoprecipitation
- DTT:
-
Dithiothreitol
- HS:
-
Heat shock
- HSFs:
-
Heat stress transcription factors
- HSPs:
-
Heat shock proteins
- IEF:
-
Isoelectric focusing
- IPG:
-
Immobilized pH gradients
- LC-ESI:
-
Liquid chromatography-electrospray ionization
- MALDI:
-
Matrix-assisted laser desorption/ionization
- NH4HCO3 :
-
Ammonium bicarbonate
- RNAi:
-
RNA interference
- SDS-PAGE:
-
Sodium dodesil sulfate-polyacrylamide gel electrophoresis
- TFA:
-
Trifluoroacetic acid
- TIFF:
-
Tag Image File Format
References
Abril N, Gion JM, Kerner R, Müller SG, Navarro CRM, Plomion C (2011) Proteomics research on forest trees, the most recalcitrant and orphan plant species. Phytochemistry. 72:1219–1242
Bose U, Broadbent JA, Byrne K, Hasan S, Howitt CA, Colgrav ML (2019) Optimization of protein extraction for in-depth profiling of the cereal grain proteome. J Proteomics 197:23–33
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Chen Q, Phoolcharoen HJ, Mason WHS (2011) Gemini viral vectors based on bean yellow dwarf virus for production of vaccine antigens and monoclonal antibodies in plants. Hum Vaccine 7:331–338
Chen X, Lin S, Liu Q, Huang J, Zhang W, Lin J, Wang Y, Ke Y, He H (2014) Expression and interaction of small heat shock proteins (sHsps) in rice in response to heat stress. Biochimica Biophysica Acta. 1844:818–828
Ehrnsperger M, Gräber S, Gaestel M, Buchner J (1997) Binding of non-native protein to Hsp25 during heat shock creates a reservoir of folding intermediates for reactivation. EMBO J 16:221–229
Feder ME, Hofmann GE (1999) Heat-shock proteins, molecular chaperones, and stress response: evolutionary and ecological physiology. Annu Rev Physiol 61:243–282
Haslbeck M, Walke S, Stromer T, Ehrnsperger M, White HE, Chen S, Saibil HR, Buchner J (1999) Hsp26: a temperature-regulated chaperone. EMBO J 18:6744–6751
Hu XI, Liu L, Song J, Zong X, Li D (2009) Over-expression of Zea mays L. protein phosphatase 2C gene (ZmPP2C) in Arabidopsis thaliana decreases tolerance to salt and drought. J Plant Physiol 166:531–542
Hu XI, Yang YF, Gong F, Zhang D, Wu L, Li C, Wang W (2015) Protein sHSP26 improves chloroplast performance under heat stress by interacting with specific chloroplast proteins in maize (Zea mays). J Proteomics 115:81–92
Irar S, Brinib F, Godaya A, Masmoudib K, Pages M (2010) Proteomic analysis of wheat embryos with 2-DE and liquid-phase chromatography a wider perspective of the proteome. J Proteomics 73:1707–1721
Klein RD, Chidawanyika T, Tims HS, Meulia T, Bouchard RA, Pett VB (2014) Chaperone function of two small heat shock proteins from maize. Plant Sci 221:48–58
Krishna P, Gloor G (2001) The Hsp90 family of proteins in Arabidopsis thaliana. Cell Stress Chaperones 6:238–246
Lee DG, Ahsan N, Lee SH, Kang KY, Bahk JD, Lee IJ et al (2007) A proteomic approach in analyzing heat-responsive proteins in rice leaves. Proteomics 7:3369–3383
Li D, Wang L, Liu X, Cui D, Chen T, Zhang H, Zhao L (2013) Deep sequencing of maize small RNAs reveals a diverse set of micro RNA in dry and imbibed seeds. PloS One 8:e55107
Magdeldin S, Enany S, Yoshida Y, Xu B, Zhang Y, Zureena Z, Yamamoto T (2014) Basics and recent advances of two-dimensional polyacrylamide gel electrophoresis. Clin Proteomics 11:166
Mahmoud FE, Rashed MA, Khalil KM, Abou-Deif MH (2018) Identification and characterization of heat shock proteins in four Egyptian maize inbred lines (Zea mays L.). Arab Universities Journal of Agricultural Sciences 26(2A):1203–1211.
Mogk A, Schlieker C, Friedrich KL, Schönfeld HJ, Vierling E, Bukau B (2003) Refolding of substrates bound to small Hsps relies on a disaggregation reaction mediated most efficiently by ClpB/DnaK. J Biol Chem 278:31033–31042
Nieto-Sotelo J, Vierling E, Ho TD (1990) Cloning, sequence analysis, and expression of a cDNA encoding a plastid localized heat shock protein in maize. Plant Physiol 93:1321–1328
Pergande MR, Cologna SM (2017) Isoelectric point separations of peptides and proteins. Proteomes 5:4–10
Rizhsky L, Liang H, Shuman J, Shulaev V, Davletova S, Mittler P (2004) When defense pathways collide: the response of Arabidopsis to a combination of drought and heat stress. Plant Physiol 134:1683–1696
Sanmiya K, Suzuki K, Egawa Y, Shono M (2004) Mitochondrial small heat-shock protein enhances thermo-tolerance in tobacco plants. FEBS Lett 557:265–268
Schuster AM, Davies E (1983) Ribonucleic acid and protein metabolism in pea epicotyls II. Response to wounding in aged tissue. Plant Physiol 73:817–821
Shevchenko A, Wilm M, Vorm O, Mann M (1996) Mass spectrometric sequencing of proteins from silver-stained polyacrylamide gels. Anal Chem 68:850–858
Sun L, Wu Y, Su S, Liu H, Yang G, Li S, Yuan Y. (2012) Differential gene expression during somatic embryogenesis in the maize (Zea mays L.) inbred line H99. Plant Cell, Tissue and Organ Culture (PCTOC) 109:271–286.
Sung DY, Vierling E, Guy CL (2001) Comprehensive expression profile analysis of the Arabidopsis Hsp70 gene family. Plant Physiol 126:789–800
Tan BC, Lim YS, Lau SE (2017) Proteomics in commercial crops: an overview. J Proteomics 169:176–188
Timperio AM, Egidi MG, Zolla L (2008) Proteomics applied on plant abiotic stresses: role of heat shock proteins (HSP). J Proteomics 71:391–411
Westermeier R (2014) Looking at proteins from two dimensions: a review on five decades of 2D electrophoresis. Arch Physiol Biochem 120:168–172
Acknowledgements
Not applicable
Funding
The study was funded by the Spanish Agency for International Development Cooperation of Spain through the research project “11-CAP-2-1151”.
Author information
Authors and Affiliations
Contributions
MH designed the study and was a major contributor in writing the manuscript. MA participated in the design of the study and reviewed the manuscript. KM participated in analysis and interpretation the data. FE carried out the experimental work of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License http://creativecommons.org/licenses/by/4.0/, which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Abou-Deif, M.H., Rashed, M.AS., Khalil, K.M. et al. Proteomic analysis of heat shock proteins in maize (Zea mays L.). Bull Natl Res Cent 43, 199 (2019). https://doi.org/10.1186/s42269-019-0251-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42269-019-0251-2