
Abstract
Background
Mucinous tubular and spindle cell carcinoma (MTSCC) is a type of renal cell carcinoma (RCC) described as an entity with low-grade and low malignant potential. This report presents a high-grade transformation of MTSCC which is an uncommon finding in this subtype of RCC. Although most reported cases showed low malignant potential and good prognosis after surgical resection, a small subset of patients has been reported with aggressive phenotype and fatal outcome.
Case presentation
A case of a 23-year-old male is discussed with an incidental renal mass in the left kidney after a workup of bilateral flank pain. Morphologically, the tumor was composed of tubular structures lined by cuboidal cells mixed with spindle cells and sarcomatoid change was present in the tumor. Previously sarcomatoid change has been mostly reported along with conventional morphology in other common type of RCC with a worse prognosis.
Conclusions
The aim of this report is to draw the attention of pathologists and clinicians to the importance of high grade MTSCC owing to the unfavorable prognosis.
Similar content being viewed by others


Background
There are various types of renal cell carcinomas with distinct underlying molecular features, unique immunophenotypes, varying degrees of clinical outcomes, and therapeutic implications. Originally, mucinous tubular and spindle cell carcinoma (MTSCC) was described as an unclassifiable tumor in 1998 (Lima et al. 2013). In 2004, it was described as a distinct entity (Yang et al. 2010). It is more commonly reported in females and the mean age at presentation is 53 years (Ferlicot et al. 2005). Although MTSCC itself has low malignant potential, rare cases with sarcomatoid change have been reported with aggressive behavior. The MTSCC is composed of spindle cells admixed with tubules lined by bland cuboidal cells set in a background of a variable amount of myxoid stroma (Ellis and Flower 2004).
We present a case of a 23-year-old male who reported bilateral flank pain. A left renal mass was detected on computerized tomography (CT) scan followed by a histopathological diagnosis of MTSCC with 70% tumor showing the sarcomatoid change.
Case presentation
A 23-year-old male reported to the urology department with chief complaints of bilateral flank pain and fever from the last fifteen days associated with a history of weight loss. There was no history of hematuria. He was a non-smoker and had no history of tuberculosis, diabetes mellitus, hypertension, asthma, or ischemic heart disease. There is no family history of any kind of malignancy. On physical examination, the patient was vitally stable with unremarkable general and systemic examination.
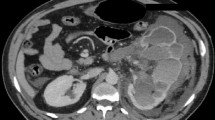
All the hematological investigations including renal function tests were within normal limits. CT scan chest showed tiny pulmonary nodules suspicious to be metastatic. CT scan (Fig. 1) abdomen showed well-defined heterogeneously relatively hypo-enhancing, ball-shaped, left renal mass with loss of fat interface with adjacent large bowel, compressing and displacing pelvicalyceal system with no definite vascular extension. A large hypodense lesion in segment VI of the liver and additional smaller hypodense lesions in the right lobe were also noted. Multiple enlarged upper abdominal and left renal hilar lymph nodes were seen.
In wake of complete clinical, laboratory, and radiological assessment left radical nephrectomy and adrenalectomy with lymph node dissection was performed.
On gross examination, a 10.5 × 9.0 × 6.0 cm well-circumscribed tumor mass was found at the lower pole with necrosis on the cut surface. The tumor was limited to the renal parenchyma, away from sinus fat. Histological analysis revealed a tumor comprised of tubular structures set in a mucinous background. The tubules were lined by cuboidal cells having highly pleomorphic nuclei. At places, the cuboidal cells merged with spindle cell areas. Almost 70% of the tumor showed sarcomatoid differentiation and tumor necrosis was also present. The sarcomatoid cells were pleomorphic with hyperchromatic nuclei and prominent nucleoli as in Fig. 2. Abundant mitotic figures were also present. The tumor was found to be focally invading into the perinephric fat, beyond the renal capsule. Carcinoma was found positive in 09 out of 13 periaortic lymph nodes.
On immunohistochemistry, cytokeratin 7 (CK7) and alpha-methyl co-A racemase (AMACR) were positive, paired-box gene 8 (PAX-8) and carbonic anhydrase IX (CA-IX) were focal positive, CD10 was focal weak positive while TFE3 was negative in the tumor as illustrated in Fig. 3. According to these morphological and immunohistochemical findings, a diagnosis of high-grade MTSCC was made.
The postoperative course of the patient was uneventful and was discharged on day 3 in a stable condition. On sixth month follow-up, the patient was in good health.

CT Scan Abdomen showing left renal mass
Discussion
MTSCC is a rare neoplasm, mostly following an indolent course, but some cases of MTSCC have been reported showing sarcomatoid change (Dhillon et al. 2009; Pillay et al. 2008; Simon et al. 2008; Isono et al. 2020; Arafah and Zaidi 2013; Sugimoto et al. 2019; Kenney et al. 2015; Gong et al. 2020). The characteristic features of seven cases are compared with our case in Table 1. Sarcomatoid change has been previously more commonly reported in other common types of renal cell carcinoma. Patients with renal cell carcinoma showing sarcomatoid change presented at an advanced stage and followed a bad prognosis (Venturina et al. 2001).
The tumor is prevalent in females with a ratio of 4:1 against males and an age group of 13 to 82 years (Yang et al. 2010; Zhao et al. 2015). MTSCC was previously considered to be originating from the loop of Henle or collecting duct but later immunohistochemical analysis showed it to be CK7 and AMACR positive. This suggested its origin from the proximal segment of the nephron showing its close relation to papillary renal cell carcinoma. Occasionally, the differentiation between both types of carcinomas becomes difficult based on routine histology and immunohistochemistry, necessitating the use of genetic studies for making a proper diagnosis (Shen et al. 2007).
Imaging studies show that the tumor mostly appears well-circumscribed and homogenous with heterogeneous and slow progressive enhancement in the corticomedullary and nephrogenic phase of CT and MRI. It has a high T2 and a low ADC value on MRI. (Cornelis et al. 2017).
On gross examination, the tumor has distinct borders with most of the tumor bulk to be found in the medullary area. Morphological analysis shows that the tumor is composed of elongated branching tubules and cords admixed with spindle cell areas. The tubular structures are lined by cuboidal cells containing scant eosinophilic cytoplasm and low-grade nuclei. Mucinous stroma is almost always present in all cases. Sometimes papillary structures, foamy macrophages, sarcomatoid change, and necrosis can be identified in tumors. The sarcomatoid cells have a high nuclear to cytoplasmic ratio, pleomorphic hyperchromatic nuclei, and prominent nucleoli (Ro et al. 1983).

A Spindle cell proliferation against myxoid stroma (B) with areas containing cords or tubules of cuboidal cells (H&E X10M), (C) foci showing higher nuclear grade and (D) sarcomatoid change (H&E X 20 M)

A CK7 (X10M), (B) AMACR (X20M), (C) CD10 (X10M) and (D) lymph node showing metastatic carcinoma (H&E X20M)
In our case, the sarcomatoid change was present constituting 70% of the tumor. In addition, 40% of the tumor was showing necrotic areas. This upgraded the tumor to ISUP grade four which shows its deviation from commonly reported cases of a low-grade type of MTSCC.
Immunohistochemically, the tumor usually shows positivity for CK7, AMACR, renal cell carcinoma (RCC) antigen, and Lewis X (CD15) in both tubular and mucinous areas. CD 10 is negative most of the time however in some cases it may be positive (Sternberg et al. 2010).
Genetic studies have shown various chromosomal abnormalities including losses of chromosomes 1, 4, 6, 8, 9, 13, 14, 15, 22 and X (Rakozy et al. 2002) and gains of chromosomes involving 2, 4, 5, 7, 9, 10, 12, 16, 17, 18, 19, 20, 22 and X (Dhillon et al. 2009; Brandal et al. 2006). Recently genetic study of MTSCC using array comparative genomic hybridization showed losses involving chromosomes 1, 4, 6, 8, 9, 13, 14, 15, 22 without any gains in chromosomes, in low and high-grade MTSCC of classical type, while the tumors having histopathological features overlapping with papillary RCC showed a variable pattern of losses and gains in chromosomes, including gains involving chromosomes 7 and 17 in 2 out of 4 analyzable tumors (Peckova et al. 2015). However, FISH based studies show that MTSCC does not show gains of chromosomes 7 and 17 (Cossu-Rocca et al. 2006). This emerging evidence has proved that although morphological and immunohistochemical features overlap with those of papillary RCC, genetically it is a separate entity.
There are no definite guidelines for the treatment of MTSCC, however surgical excision remains adequate for localized cases. For metastatic tumors, improvement in patient survival and reduced tumor burden have been achieved in some cases by benefiting from the use of sunitinib, a tyrosine kinase inhibitor (Volpe et al. 2011). In a recent case report, combined immunotherapy with ipilimumab and nivolumab showed complete recession of the metastatic MTSCC (Fuchizawa et al. 2021). In a study (Lebacle et al. 2019), renal cell carcinomas with sarcomatoid change showed better expression of PD-1/PD-L1 as compared to conventional carcinomas. So, sarcomatoid RCC will respond better to targeted immunotherapy. However, further studies are required for setting proper management plan for metastatic MTSCC.
Conclusions
MTSCC is a rare tumor. This case report presents rare renal carcinoma subtype with sarcomatoid change. The sarcomatoid change is associated with aggressive behavior which is why the identification of this high-grade feature is essential. Complete morphology, immunohistochemistry and cytogenetics are required for definite diagnosis of this tumor. Close follow-up of tumor is required for understanding of complete biological behavior of this tumor.
Availability of data and materials
The datasets generated and/or analyzed during the current study are not publicly available due to hospital privacy policy but can be made available from the corresponding author on reasonable request and after approval from IRB & EC.
Abbreviations
- MTSCC:
-
Mucinous tubular and spindle cell carcinoma
- RCC:
-
Renal cell carcinoma
- CK-7:
-
cytokeratin 7
- CT:
-
Computerized tomography
- MRI:
-
Magnetic resonance imaging
- CA-IX:
-
Carbonic anhydrase IX
- AMACR:
-
alpha methyl co-A racemase
References
Arafah M, Zaidi SN. Mucinous tubular and spindle cell carcinoma of the kidney with sarcomatoid transformation. Saudi J Kidney Dis Transpl. 2013;24(3):557–60.
Brandal P, Lie AK, Bassarova A, Svindland A, Risberg B, Danielsen H. Genomic aberrations in mucinous tubular and spindle cell renal cell carcinomas. Mod Pathol. 2006;19(2):186–94.
Cornelis F, Ambrosetti D, Rocher L, et al. CT and MR imaging features of mucinous tubular and spindle cell carcinoma of the kidneys. A multi-institutional review. Eur Radiol. 2017;27(3):1087–95.
Cossu-Rocca P, Eble JN, Delahunt B, Zhang S, Martignoni G, Brunelli M, et al. Renal mucinous tubular and spindle carcinoma lacks the gains of chromosomes 7 and 17 and losses of chromosome Y that are prevalent in papillary renal cell carcinoma. Mod Pathol. 2006;19(4):488–93.
Dhillon J, Amin MB, Selbs E, Turi GK, Paner GP, Reuter VE. Mucinous tubular and spindle cell carcinoma of the kidney with sarcomatoid change. Am J Surg Pathol. 2009;33(1):44–9.
Ellis SM, Flower C. (2004) WHO Classification of Tumours Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs. 1st Edition.
Ferlicot S, Allory Y, Compe ́ rat E. Mucinous tubular and spindle cell carcinoma: A report of 15 cases and a review of the literature. Virchows Arch. 2005;447:978–83.
Fuchizawa H, Kijima T, Takada-Owada A, et al. Metastatic mucinous tubular and spindle cell carcinoma of the kidney responding to nivolumab plus ipilimumab. IJU Case Rep. 2021;4(5):333–7.
Gong P, Zhuang Q, Wang X, et al. Mucinous tubular and spindle cell carcinoma of the kidney: Five case reports and review of the literature. Oncol Lett. 2020;20(6):337.
Isono M, Seguchi K, Yamanaka M, et al. Rapid progression of mucinous tubular and spindle cell carcinoma of the kidney without sarcomatoid changes: A case report, Urol. Case Rep. 2020;31:101162.
Kenney PA, Vikram R, Prasad SR, et al. Mucinous tubular and spindle cell carcinoma (MTSCC) of the kidney: a detailed study of radiological, pathological and clinical outcomes. BJU Int. 2015;116(1):85–92.
Lebacle C, Pooli A, Bessede T, Irani J, Pantuck AJ, Drakaki. A. Epidemiology, biology and treatment of sarcomatoid RCC: current state of the art. World J Urol. 2019;37(1):115–23.
Lima MS, Barros-Silva GE, Pereira RA, et al. The imaging and pathological features of a mucinous tubular and spindle cell carcinoma of the kidney: a case report. World journal of surgical oncology. 2013;11(1):34.
Peckova K, Martinek P, Sperga M, et al. Mucinous spindle and tubular renal cell carcinoma: analysis of chromosomal aberration pattern of low-grade, high-grade, and overlapping morphologic variant with papillary renal cell carcinoma. Annals of Diagnostic Pathology. 2015;19(4):226–31.
Pillay N, Ramdial PK, Cooper K, Batuule D. Mucinous tubular and spindle cell carcinoma with aggressive histomorphology--a sarcomatoid variant. Hum Pathol. 2008;39(6):966–9.
Rakozy C, Schmahl GE, Bogner S, Storkel S. Low-grade tubular-mucinous renal neoplasms: morphologic, immunohistochemical, and genetic features. Mod Pathol. 2002;15(11):1162–71.
Ro JY, Ayala AG, Sella A, et al. Sarcomatoid renal cell carcinoma: clinicopathologic. A study of 42 cases. Cancer. 1983;130:657–9.
Shen SS, Ro JY, Tamboli P. Mucinous tubular and spindle cell carcinoma of the kidney is probably a variant of papillary renal cell carcinoma with spindle cell features. Ann Diagn Pathol. 2007;11(1):13–21.
Simon RA, di Sant’agnese PA, Palapattu GS, et al. Mucinous tubular and spindle cell carcinoma of the kidney with sarcomatoid differentiation. Int J Clin Exp Pathol. 2008;1(2):180–4.
Sternberg SS, MillsStacey E, Carter D. Sternberg’s Diagnostic Surgical Pathology. 5th ed. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2010.
Sugimoto R, Uesugi N, Yamada N, et al. Sarcomatoid change associated with epithelial-mesenchymal transition in mucinous tubular and spindle cell carcinoma of the kidney: a case report. Int J Clin Exp Pathol. 2019;12(7):2767–71.
Venturina MP, Moch H, Amin M, et al. Sarcomatoid differentiation in renal cell carcinoma. Am J Surg. 2001;25:275–84.
Volpe A, Cadeddu JA, Cestari A, Gilld IS, Jewett MA. Con13. temporary management of small renal masses. Eur Urol. 2011;60:501–15.
Yang G, Breyer BN, Weiss DA, MacLennan GT. Mucinous tubular and spindle cell carcinoma of the kidney. The Journal of urology. 2010;183(2):738.
Zhao M, He XL, Teng XD. Mucinous tubular and spindle cell renal cell carcinoma: a review of clinicopathologic aspects. Diagn Pathol. 2015;10:168.
Acknowledgements
Not applicable.
Funding
The authors have not received any funding from any source for the publication of this article.
Author information
Authors and Affiliations
Contributions
NH: conception, acquisition, analysis, interpretation of data; has drafted the work and has approved the submitted version. AM: supervision, revised, analysis, interpretation of data; and has approved the submitted version. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The research has been approved by the Institutional Review Board and Ethics Committee (IRB & EC), Shifa International Hospitals Limited.
Consent for publication
The manuscript does not contain data from any person, and hence the above statement is “Not applicable”.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hasan, N., Moatasim, A. Mucinous tubular and spindle cell carcinoma with high-grade transformation: case report. Surg Exp Pathol 5, 4 (2022). https://doi.org/10.1186/s42047-022-00105-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42047-022-00105-x