
Abstract
Background
Glioblastoma multiform with primitive neuronal component (GBM-PNC) has been recently defined as a rare variant of glioblastoma multiform (GBM), which shows characteristically pathological pattern of less differentiated areas with small blue cell morphology and neuroectodermic immunophenotype. New studies emphasize its characteristics and differences, which have become vitally important due to the changes in therapeutic management.
Case presentation
We present the case of 57-year-old male patient who onset symptoms were secondarily widespread partial seizures and expression aphasia. Brain magnetic resonance imaging (MRI) reported left enhanced temporal infiltrating lesion, requiring surgery twice throughout two years. At first surgery, pathological samples revealed embryonic tumor of the central nervous system (grade IV, WHO 2016), so PACKER protocol consisting of CSRT (craniospinal radiation) plus weekly vincristine followed by 8 cycles of cisplatin, lomustine and vincristine usually used for medulloblastomas or other primitive neuroectodermal tumors was started. However, due to reappearance of symptoms and progression in MRI, reoperation was performed with definitive diagnosis of GBM-PNC (Grade IV, WHO 2016) and switched to STUPP protocol.
Conclusions
It is important to take into account the chance of this entity when histological, radiological and intraoperative findings orient toward a primitive neural tumor since the presence of GBM could be overlooked leading to mistakes in diagnosis and the therapeutic orientation.
Similar content being viewed by others


Background
In 2016 the classification of WHO tumors of the central nervous system (CNS) made changes of nomenclature appearing the new description of glioblastoma with primitive neuronal component (GBM-PNC) replacing the previous term of glioblastoma multiform-primitive neuroectodermic tumor (GBM-PNET) [1]. GBM is the most common malignant primary brain tumor in the CNS. Advances in molecular biology and cytogenetic allowed to identify the variant with primitive neural component, which is only 0.5% of the total GBM [2]. These tumors occur predominantly in adults with an average of 54 years old [3]. GBM-PNC is a unique variant of GBM at molecular level with high frequency of TP53, PIK3CA, PIK3R1 or PTEN mutation [4]. Unlike GBM, primitive neuronal component (PNC) tumor has a better response to therapy and higher survival rate of approximately 38% in 4 years, although it has an increased risk of spread to cerebrospinal fluid with extracranial metastases [5,6,7]. Its treatment differs from GBM protocol, because after surgical resection needs craniospinal irradiation and platinum base land chemotherapy to prevent dissemination [8].
Case presentation
We present the case of a 57-year-old male patient with no pathological history of interest that made his debut with an expression aphasia plus behavioral alteration; later in the emergency room, he suffered a secondarily widespread partial seizure. MRI found a left temporal lesion with small areas of contrast enhancement in the T1 gadolinium with an increase in FLAIR component (Fig. 1). The anatomic–pathological study of the first surgical resection evidenced a embryonic tumor of the central nervous system, B-catenin positive, INI-1 not mutated, P53 < 10%, Ki67 95%, IDH-1 negative, ATRX not mutated. The diagnostic challenge was mainly to identify the correct diagnosis because in this case, at first surgery the high-grade glial component was not identified. The first operation obtained a rough resection of the > 95%. Later, he started oncological specific treatment with 4 cycles of PACKER protocol [9] for PNET and then cranial–spinal radiotherapy with 36 gy plus boost in the tumoral area with 55 gy. During treatment, the patient presented again speech disturbance and right hemiparesis. The new MRI reported tumor growth with contrast enhancing and marked increase of the FLAIR component (Fig. 2). Given the tumor progression, the patient went to surgical reintervention 7 months after the first surgery, where this time anatomic–pathological diagnosis concluded GBM-PNC (Grade IV, WHO 2016). In the second pathology sample, a GBM-PNC is evidenced, IDH-1 and IDH-2 not mutated, MGMT not methylated, 1p19q not codeleted, NEuN negative, Olig-2 focally positive < 25%, high-grade neuroembrional and glial component [10]. Intraoperatively, macroscopic differences in neoplasm tissues were evident. At first surgery, dura mater appeared infiltrated, the tumor had malignant aspect, and no tumoral angiogenesis was present. Otherwise, necrosis was not found. The consistency was soft and easily removable with aspirator (Fig. 3). At second surgery, the tumoral tissue was hard and looked malignant, it was difficult to aspirate, and it had intense positive response to 5-aminolevulinic acid (5-ALA). Subsequently, treatment was changed to STUPP protocol. Patient finally died 14 months after first surgery.

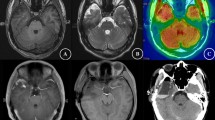
Brain MRI T1 with contrast axial cut (A) and coronal (B) visualizing contrast-capturing left temporal lesion (red arrows)

MRI cerebral axial cuts (A and B) and coronal (C) displaying increased FLAIR component (yellow circle) and contrast uprising lesion (red arrows)

A HE × 40 embryonic undifferentiated component, blue cell, densely cellular, pseudo-red shaper. B HE × 40 astrocytic glial component with moderate atypia. C Intraoperative image visualizing embryonic component of the first surgery (arrow)
Discussion
Histologically, the presentation of this tumor is usually characterized by the high expression of fibrillary glial protein (GFAP) in the GBM areas and focal to patchy in the undifferentiated hypercellular areas of PNET. Perry et al. described infiltrative growth pattern with commonly secondary structures of Scherer associated. Additionally, at low magnification the PNET component looks like markedly hypercellular (small blue cell); nodules with features of high nuclear to cytoplasmic ratios, hyperchromatic oval to carrot-shaped nuclei, high mitotic-karyorrhectic indices and Homer Wirth (neuroblastic) rosettes principally [11]. Immunohistochemically, it presents neuronal immunophenotype (S-100, Synaptophysin, NeuN, NSE and NFP) and evidence of multiple areas of hypercellular PNET [11]. Although genetic alterations may be encountered, glioma like characteristics such as 10q deletion, EGFR amplification, 1p/19q deletions and a high Ki 67 set with N-myc and C-myc extended may be observed [12]. The IDH1 and IDH2 are still controversial [13].
Typically, this tumor had high rate of temporal locations [14]. MRI radiological features of this tumor reveal usually a T2 heterogeneous mass, a T1 gadolinium showing a well-circumscribed lesion with vasogenic edema, heterogeneous enhancement and may be associated with central necrosis, cyst formations and tumoral hemorrhage [6, 15,16,17]. Nowadays, spectroscopy is not useful because results show a malignancy pattern tumor with high peak of choline and lactate with decreases of N-acetyl aspartate, but not specific features that might help with diagnosis [15].
The 2016 WHO CNS tumor nomenclature changed to respond the complex situation that was generated when presentation of GBM-PNET was discovered [1]. How to perform the treatment approach to these patients became a challenge, because misdiagnosing such as the case we present may occur. This may lead to an initial histological PNC tumor treatment with protocol PACKER and focus on avoiding the dissemination to cerebrospinal fluid, but without treatment for the high glial neoplasm [17]. The controversy in this case appeared in first surgery when macroscopical features suggest malignancy and pathologist confirmed the absence of GBM tissue compatibility. Despite knowing that PNET tumors alone are extremely rare at this patient's age, taking into account that the surgical resection of the tumor was almost complete, and that pathology reported no GBM tissue, the oncologist made the decision to treat as a PNET neoplasm. Retrospectively, we think that the peripheral tissue near to eloquent areas which we decided not to remove due to high chances of postoperative deficit, really was the GBM tissue compatible that later grew up.
Control MRI showed progression of the neoplasm with contrast enhancement, which is very atypical for this type of tumor. Therefore, the patient required a second surgery with evidence of macroscopical, 5-ALA enhancement and histological GBM features, immediately switching the oncological treatment to STUPP protocol. We concluded that highly probable the initial treatment was suboptimal, leading to the fast progression and compromised the patient's evolution allowing the high-grade glial component to grow. The average survival of these patients is a little better than GBM [11]. Without the ideal approach to their treatment at the beginning, the prognosis and evolution may be affected and consequently is very important in the initial diagnosis of PNC tumors to analyze the surgical part in detail because the high-grade glial component may be hidden [3]. Additionally, like in this case, there should be the high suspicious of first surgery pathology misdiagnosis, taking into account the age of the patient and epidemiology. We suggest a closer MRI follow-up in the postoperative phase to detect early unexpected tumor grows up and the use of 5-ALA protocol in centers where is possible to perform initial surgery.
Since there is no treatment protocol established for GBM-PNC and the survival studies for this entity are unclear, the diagnosis and continuous treatment is a challenging area, taking the clinicians to many controversies and differences to resolve [14]. However, as GBM-PNC has recently been shown to have more PNC clinical behavior with increased risk of CSF spread, there could be a platinum-based chemotherapy benefit after GBM treatment has failed [8]. Given the weirdness of this presentation, and the importance to have on mind the differential diagnosis and approach, we present a clinical case with histopathological, radiological and intraoperative features.
Conclusions
The variant of GBM-PNC causes a biphasic component in anatomical-pathological preparations. When the blue small cell component predominates, it makes difficult to differentiate with other blue small cell tumors (CNS embrionary tumor, small cell carcinoma metastasis and lymphoma) or mixed tumors, leading to mistakes in the diagnosis affecting the treatment and prognosis of the patient. This rare variant of GBM needs to be considered to avoid misdiagnosis.
Availability of data and material
Not applicable.
Abbreviations
- GBM-PNC:
-
Glioblastoma multiform with primitive neuronal component
- MRI:
-
Magnetic resonance imaging
- CNS:
-
Central nervous system
- GBM-PNET:
-
Glioblastoma multiform-primitive neuroectodermic tumor
- GFAP:
-
Fibrillary glial protein
- CSRT:
-
Craniospinal radiation
- 5-ALA:
-
5-Aminolevulinic acid
References
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella D, Webster B, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131:803–20.
Papadopoulos EK, Fountas KN, Brotis AG, Paterakis KN. A supratentorial primitive neuroectodermal tumor presenting with intracranial hemorrhage in a 42-year-old man: a case report and review of the literature. J Med Case Rep. 2013;7:1–6.
Borys E, Prabhu VC, Pambuccian SE. Glioblastoma with primitive neuronal component: cytologic findings in intraoperative squash preparations. Diagn Cytopathol. 2019;47(3):234–7.
Xu G, Zheng H, Li JY. Next-generation whole exome sequencing of glioblastoma with a primitive neuronal component. Brain Tumor Pathol. 2019;36(3):129–34. https://doi.org/10.1007/s10014-019-00334-1.
Tamai S, Kinoshita M, Sabit H, Furuta T, Miyashita K, Yoshimura K, et al. Case of metastatic glioblastoma with primitive neuronal component to the lung. Neuropathology. 2019;39(3):218–23.
Georgiu C, Mihuţ E, Raus I, Mirescu ŞC, Szabo L, Şovrea AS. Pediatric glioblastoma with giant cells and “supratentorial” primitive neuroectodermal component: case report and review of the literature. Rom J Morphol Embryol. 2015;56(3):1165–71.
Kay MD, Pariury HE, Perry A, Winegar BA, Kuo PH. Extracranial metastases from glioblastoma with primitive neuronal components on FDG PET/CT. Clin Nucl Med. 2020;45(3):E162–4.
Prelaj A, Rebuzzi SE, Caffarena G, Berrìos JRG, Pecorari S, Fusto C, et al. Therapeutic approach in glioblastoma multiforme with primitive neuroectodermal tumor components: case report and review of the literature. Oncol Lett. 2018;15(5):6641–7.
Greenberg HS, Chamberlain MC, Glantz MJ, Wang S. 11305414. Neuro Oncol. 2001;3:29–34.
Karsy M, Gelbman M, Shah P, Balumbu O, Moy F, Arslan E. Established and emerging variants of glioblastoma multiforme: review of morphological and molecular features. Folia Neuropathol. 2012;50(4):301–21.
Perry A, Miller CR, Gujrati M, Scheithauer BW, Zambrano SC, Jost SC, et al. Malignant gliomas with primitive neuroectodermal tumor-like components: a clinicopathologic and genetic study of 53 cases. Brain Pathol. 2009;19(1):81–90.
Brown TJ, Brennan MC, Li M, Church EW, Brandmeir NJ, Rakszawski KL, et al. Association of the extent of resection with survival in glioblastoma a systematic review and meta-Analysis. JAMA Oncol. 2016;2(11):1460–9.
Karsy M, Huang TKG. Molecular, histopathological, and genomic variants of glioblastoma. Front Biosci. 2017;19:1065–87.
Onak Kandemir N, Bahadir B, Gul S. Glioblastoma with primitive features: case report. 2009;260–4.
Kim DG, Lee DY, Paek SH, Chi JG, Choe G, Jung HW. Supratentorial primitive neuroectodermal tumors in adults. J Neurooncol. 2002;60(1):43–52.
Donabedian P, Tuna I, Rahman M, Gregory J, Kresak J, Rees JH. Glioblastoma with a primitive neuroectodermal component: two cases with implications for glioblastoma cell-of-origin. Clin Imaging. 2020;2021(73):139–45. https://doi.org/10.1016/j.clinimag.2020.10.041.
Forbes V, Vredenburgh J. Primitive neuroectodermal tumor with glioblastoma multiforme components in an adult: a collision tumor. Cureus. 2016;8(1):1–7.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
SV helped in conception, design of the work, analysis, writing of the paper. AO was the neurosurgeon of the patient in the two surgeries and helped in acquisition of original images including intraoperative and radiological images. MC was the pathologist of the case, histological diagnosis and was involved in acquisition of histological images and examples of the case. JR contributed to revision of all manuscripts, expert advices and corrections of the paper, theoretical analysis of discussion. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Yes, a written informed consent to publish was obtained from a study participant.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Valbuena, S., Ortega, A., Centeno, M. et al. Glioblastoma multiform with primitive neuronal component, radiological and histology features: a case report. Egypt J Neurosurg 36, 37 (2021). https://doi.org/10.1186/s41984-021-00135-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41984-021-00135-9