
Abstract
Introduction
Tauopathies are a class of neurodegenerative disorders characterized by the abnormal accumulation of hyperphosphorylated tau protein in the brain. Subacute sclerosing panencephalitis (SSPE) caused by a latent aberrant measles virus infection, is characterized by extensive inflammation and neuronal impairment. A prominent pathological hallmark of SSPE described recently is the accumulation of abnormal tau protein possibly resulting from diffuse brain inflammation triggered by measles virus infection.
Short summary
Understanding the role of tau pathophysiology in SSPE is crucial for improving the diagnosis and management of this debilitating condition. Current research suggests that persistent measles virus infection in the brain leads to chronic inflammation, which in turn triggers abnormal tau phosphorylation and accumulation. Further elucidating the precise mechanisms linking measles virus infection, neuro-inflammation, and tauopathy in SSPE is essential for developing targeted therapies.
Conclusion
This narrative review provides valuable insights for both researchers and clinicians in understanding the pathological mechanisms underlying SSPE which is crucial for developing effective treatment strategies. These might include antiviral drugs to combat persistent infection, anti-inflammatory agents to reduce neuro-inflammation, or even treatments targeting tau pathology directly. Collaborative efforts among researchers, clinicians, and public health authorities are crucial for advancing our understanding of SSPE to combat this devastating disorder.
Similar content being viewed by others


Introduction
Tauopathies encompass a range of neurodegenerative diseases characterized by the accumulation of abnormal tau protein in the brain. This aberrant tau protein forms tangles within nerve cells, disrupting their function and ultimately leading to cell death. These conditions can manifest in various ways, including SSPE, Alzheimer's disease, front temporal dementia, progressive supranuclear palsy, and corticobasal degeneration, among others. Each tauopathy has its own unique clinical features and progression, but they all share this common underlying pathology of tau accumulation in the central nervous system [1]. On basis of western blotting and immunohistochemistry, tauopathies are divided into three groups. These groups are 3-repeat (3R), 4-repeat (4R) and a mix of 3-repeat and 4-repeat (3R + 4R) tauopathies. Representative 4R tauopathies include globular glial tauopathy, corticobasal degeneration, argyrophilic grain disease, and progressive supranuclear palsy (PSP). These conditions are characterized by the accumulation of both 3-repeat (3R) and 4-repeat (4R) tau isoforms. On the other hand, 3R tauopathies predominantly involve the 3-repeat tau isoforms. Examples include Pick's disease (also known as front temporal dementia with Pick bodies) and myotonic dystrophy. Chronic traumatic encephalopathy (CTE), primary age-related tauopathy (PART), and Alzheimer's disease (AD) are all also considered tauopathies, which are a group of neurodegenerative diseases characterized by the abnormal aggregation of tau protein in the brain [2]. The presence of neurofibrillary tangles (NFTs), which are abnormal aggregations of tau protein, in the brain tissues of SSPE patients suggests a potential link between SSPE and tau pathology. Moreover, the detection of aberrant tau protein in the cerebrospinal fluid (CSF) further supports this association and might serve as a biomarker for SSPE diagnosis or disease progression monitoring. Studies have indeed shown the presence of measles virus (MV) in neurons and oligodendrocytes in the brains of individuals with SSPE. These findings suggest that the virus persists in these cells and may contribute to the pathophysiology of the disease. Oligodendrocytes are particularly important because they produce myelin, the fatty substance that insulates nerve fibers and facilitates the transmission of nerve impulses. Damage to oligodendrocytes and myelin is believed to play a role in the neurological symptoms observed in SSPE [3]. However, there are limited reports discussing the role of tauopathies in SSPE patients who have received treatment. Figure 1 represents an overview of the narrative review in a glimpse.

Overview of the narrative review in one glimpse
Tau protein
Tau protein is a microtubule-associated protein that is primarily found in neurons in the central nervous system. Its main physiological role is to stabilize microtubules, which are crucial for the structural integrity and transport of neuronal cells [4]. However, when tau protein undergoes abnormal changes, it can play a pathological role in various neurodegenerative diseases, particularly in Alzheimer's disease, but also in other conditions such as frontotemporal dementia [5]. In disorders like SSPE, tau proteins become hyperphosphorylated, leading to their abnormal aggregation into neurofibrillary tangles (NFTs) and neuropil threads [6].
Subacute sclerosing panencephalitis
Subacute sclerosing panencephalitis (SSPE) is a progressive and catastrophic neurological disorder that is caused by persistent defective measles virus [7]. It is marked by the loss of neurons, atrophy affecting both gray and white matter, infiltration of inflammatory cells, and demyelination [8]. Approximately 20% of individuals with SSPE exhibit neurofibrillary tangles, which are mainly comprised of microtubule-associated protein tau that has undergone abnormal phosphorylation [9]. While tau normally stabilizes microtubule structures, the hyperphosphorylation of tau is associated with the formation of neurofibrillary tangles and subsequent neuronal degeneration [10]. The latency period from acute measles infection to the initial symptoms of SSPE is typically reported to be between 4 and 10 years, with a range spanning from 1 month to 27 years [11]. Despite a significant reduction in the disease's incidence in the post-vaccination era, it remains higher in developing countries. Saha et al. documented an annual incidence of 21 cases per million populations in India, contrasting with 2.4 cases per million populations in the Middle East [12]. The clinical progression is marked by a gradual decline in cognitive function and behavioural changes, followed by focal or generalized seizures, along with myoclonus, ataxia, visual disturbances, and eventually a vegetative state. Presently, there is no cure for SSPE, and the consensus is that its effective prevention through vaccination programs is more advantageous and cost-effective than other high-level forms of control [13].
Analysis of neurological findings, blood, cerebrospinal fluid, electroencephalograms, and imaging tests, as well as clinical symptoms, are the primary methods used to diagnose SSPE [14]. Dyken’s criteria is used for diagnosis, which include two major and four minor criteria. Major criteria include elevated anti‑measles antibody titres in CSF or ratio in serum and typical or atypical clinical history. Minor criteria include the following: (i) characteristic electroencephalogram (EEG) findings; (ii) CSF globulin levels of the total CSF protein; (iii) brain biopsy; and (iv) molecular diagnostic test showing identified mutations of wild‑type measles virus. Two major criteria plus one minor criterion is required [15]. Magnetic resonance imaging (MRI) findings in SSPE patients are not specific and may be normal in the early stage. In the early stages, parieto‑occipital and frontal involvement are more common. The involvement of the thalamus, basal ganglia and the corpus callosum is present in a significant number of patients. The cerebellum and brainstem are rarely involved [16]. Abnormal MRI findings are significantly more frequent in the later disease stages, with the thalamus, corpus callosum, and basal ganglia usually affected after the cortex has already shown signs of disease [11]. Here, EEG and MRI findings of a patient with SSPE are represented. VEEG showed generalized periodic discharges suggestive of SSPE (Fig. 2). On MRI, multiple T2/FLAIR hyper intensities was seen in bilateral frontal and occipital and sub cortical area (Fig. 3).

EEG of the patient with SSPE showing background slowing and generalized periodic discharges occurring at intervals of 7–8 s in longitudinal bipolar montage

T2/FLAIR hyperintensities in parieto-occipital region
Pathological role of Tau Protein
Neurons, with intricate morphology forming axons and dendrites for neural transmission, can undergo structural alterations that impact behaviour and lead to pathological events. The morphological transformation of neurons involves significant cytoskeletal rearrangement crucial for maintaining cell shape [17]. Tau is a hydrophilic protein that appears as a random coiled protein, primarily located in neurons and, to a lesser extent, in glial cells, mainly within axons in mature neurons. However, it is also present in the mitochondria, nucleus, plasma membrane, dendrites, and synapses, suggesting potential roles beyond its main function in microtubule regulation [18]. Cytoskeleton protein aggregation destabilizes neurons, causing structural changes that hinder the seamless transport of biomolecules in both anterograde and retrograde directions along axons. This process can obstruct axonal pathways, leading to neuronal degeneration from the lesion site back to the perikaryon. In tau-related conditions affecting structure and function, tau proteins may translocate to the cell body and dendrites [19]. The abnormal deposition of modified tau proteins in neurons is a common feature in neurodegenerative disorders known as "tauopathies" [20]. Abnormal phosphorylation of tau protein oligomers leads to the formation of neurofibrillary tangles in neurons [21]. These tangles are composed of hyperphosphorylated tau, which aggregates into twisted filaments [22]. NFTs disrupt the normal cytoskeletal structure of neurons, leading to cell dysfunction and, ultimately, neuronal death. When tau becomes hyperphosphorylated, it detaches from microtubules and loses its ability to stabilize them. This disruption of microtubule function impairs axonal transport, which is crucial for the proper delivery of essential molecules and organelles within neurons. As a result, neurons can no longer function properly, leading to cognitive and motor impairments [23]. Aberrant tau protein can spread from neuron to neuron in a prion-like manner. This can contribute to the progressive nature of tauopathies, as the misfolded tau protein can propagate and induce pathological changes in healthy neighbouring neurons [24]. Recent studies have postulated that the hyperphosphorylation of tau protein, especially on serine and threonine residues, is implicated in its aggregation [10]. Furthermore, various post-translational modifications (PTMs) such as phosphorylation, acetylation, ubiquitination, methylation, oxidation, sumoylation, O-GlcNAcylation, N-glycosylation, and cleavage have been observed in tau protein [25, 26]. These post-translational modifications (PTMs) have the potential to modify the charge, hydrophobicity, and conformation of tau, thereby affecting its function, protein–protein interactions, and aggregation. Consequently, alterations in PTMs are considered crucial indicators in the pathogenesis of neurodegenerative diseases [27, 28]. Compared to non-aggregated tau protein, aggregated forms exhibit increased resistance to dephosphorylation by protein phosphatase 2A (PP2A), contributing to tau pathology [29]. Tau, primarily an intracellular protein, is released into the extracellular space following axonal degeneration, neuronal death, or direct translocation from the cytoplasm to the plasma membrane [18]. Translocation may occur via presynaptic vesicle secretion, exosomes, ectosomes, or in its unbound state [30]. Once released, tau interacts with low-density lipoprotein receptor-related protein-1, heparan sulphate proteoglycans, or muscarinic receptors, and is internalized by neighbouring neurons through dynamin-mediated endocytosis and tunnelling nanotubes connecting cytoplasmic content between cells [31]. Once seeded, it prompts the aggregation of natively folded tau proteins within naive cells, inducing cellular toxicity and disease spread, reminiscent of a prion-like propagation hypothesis [18, 24]. Phosphorylation is widely acknowledged as a pivotal factor in regulating tau function and is closely associated with the advancement of tauopathies. The hyperphosphorylation of tau can result in its detachment from microtubules (MTs), impeding its capacity to facilitate MT polymerization. In a tauopathy mouse model, inhibiting exosome production and reducing microglia activation effectively halts the propagation of abnormal tau protein. These findings underscore the crucial role of microglia in tauopathy spread through phagocytosis and the release of exosomes carrying tau protein, suggesting that targeting microglia may represent a novel avenue for tauopathy therapies [32]. Microglia respond to inflammatory stimuli by producing pro-inflammatory mediators such as IL-1β, IL-6, and TNF-α, heightening the activity of kinases implicated in tau protein phosphorylation and exacerbating the disease [33].
Genes: a key player in the pathological responses in SSPE
SSPE is believed to result from a persistent infection of the brain by a defective form of the measles virus. This form of the virus is thought to evade the immune system and gradually cause neurological damage. The measles virus in SSPE patients undergoes mutations that allow it to persist and replicate in the brain. These mutations may render the virus less susceptible to the immune response and antiviral treatments. The immune response to the persistent measles infection in the brain is thought to play a role in the pathogenesis of SSPE. However, the exact nature of this response and how it contributes to the disease are not well understood. Genetic factors may also play a role in SSPE susceptibility or progression. Certain genetic variations may influence the immune response to the measles virus or the ability of the virus to persist in the brain. It is generally accepted that SSPE is associated with wild-type measles virus infections rather than vaccine strains. This is supported by epidemiological studies showing that SSPE occurs more frequently in regions with lower vaccination coverage and by genetic studies that have identified specific viral mutations associated with SSPE cases. However, it is important to note that the measles vaccine is highly effective at preventing measles infection and subsequent SSPE. Vaccination programs have significantly reduced the incidence of both measles and SSPE in many parts of the world [34]. Research into the exact mechanisms of SSPE is ongoing, and while mutated variants of the measles virus may play a role in the persistence of the virus within the brain, they are not typically referred to as a distinct "SSPE virus." The term "SSPE virus" might be a misnomer or an oversimplification of the complex relationship between the measles virus and the development of SSPE. Exactly! Measles virus, scientifically known as Measles morbillivirus, is indeed a member of the Morbillivirus genus within the Paramyxoviridae family and the order Mononegavirales. It is characterized by its negative-sense single-stranded RNA genome, surrounded by an envelope derived from the host cell membrane, and containing a nucleocapsid core. This virus encodes both structural proteins, which form the virus particle, and non-structural proteins involved in various aspects of the viral life cycle [35]. Pleomorphic virus particles, with an average size of 150–300 nm and a maximum size of 900 nm, are produced by this encapsulated virus. Six structural proteins are encoded by its genome, which is a single-stranded RNA consisting of 15,894 nucleotides: the polymerase (large, L) protein, the phosphorylated (P) protein, the matrix (M) protein, the fusion (F) protein, the hemagglutinin (H) protein, and the nucleocapsid (N) protein. The non-structural proteins produced by the P gene, V and C, often play roles in immune evasion and modulation, affecting how the virus interacts with the host immune system. These proteins can influence the host's sensitivity and responsiveness to the viral infection. Three proteins-phosphoprotein (P), large protein (L), and nucleoprotein are present in the inner nucleocapsid (N).The viral RNA-dependent RNA polymerase, consisting of the L and P proteins, plays a crucial role in replicating the viral genome and producing new viral RNA. The M protein is involved in virus particle formation and interacts with the RNA complex, as well as the cytoplasmic tails of the H and F proteins, which are important for viral entry into host cells. This intricate interplay between viral proteins is essential for the successful replication and spread of the measles virus [36]. The H protein (hemagglutinin) facilitates the entry of the virus into the target cell by binding to specific receptors on the cell surface. This binding event triggers the activation of the F protein (fusion protein), which allows the virus to fuse with the host cell membrane, facilitating entry into the cell. Additionally, the proteins N (nucleoprotein), P (phosphoprotein), and L (large protein) are essential for the replication of the measles virus. These proteins are involved in various stages of viral replication, including transcription and replication of the viral genome. The V and C proteins are non-structural proteins encoded by the P gene of the measles virus. These proteins play roles in modulating the host immune response and interfering with cellular processes to facilitate viral replication and spread. Furthermore, the measles virus exhibits genetic diversity, with 24 genotypes and eight genetic clades (A-H) identified for the wild-type virus. This diversity reflects the evolution and spread of the virus over time and across different geographic regions (Fig. 4) [37]. One of the main features of the SSPE virus is its inability to produce viral particles efficiently, which is thought to contribute to its persistence in the brain. The genetic alterations, particularly in genes such as M (matrix), H (hemagglutinin), N (nucleocapsid), and F (fusion), likely play a role in this impaired viral replication and altered protein expression. The compromised envelope protein expression in the SSPE virus likely contributes to its ability to evade the immune response and establish persistent infection in the brain's neural cells. This persistence leads to the progressive neurological deterioration characteristic of SSPE [38]. Genetic investigations alongside epidemiological studies have consistently shown that the measles vaccine virus does not lead to SSPE. The primary reason for this is the rapid clearance of the vaccine virus by the host's immune system after vaccination. This clearance prevents the virus from persisting in the body and causing the neurological complications associated with SSPE. This reinforces the critical importance of widespread measles vaccination in preventing not only measles, but also its potential complications like SSPE. Absolutely, the interplay between genetic factors and viral persistence can be quite intricate. In the case of subacute sclerosing panencephalitis (SSPE), which is a rare but severe complication of measles virus infection, certain genetic variations can indeed affect how the immune system responds to the virus. These variations, such as single nucleotide polymorphisms (SNPs) in genes related to the immune system, can potentially influence the likelihood of persistent infection and the development of SSPE. SNPs are variations in a single nucleotide occurring at a specific position in the genome and can have various effects on gene function and expression. Genes associated with acquired immunity, such as IL2, IL12, IL4, IL17, IL18, IL22, IL23, Interferon-gamma, GZMB, and PD1, play crucial roles in adaptive immune responses, including T cell activation, cytokine production, and immune regulation. On the other hand, genes linked to innate immunity, such as TLR3 (Toll-like receptor 3), TLR4 (Toll-like receptor 4), MxA (Myxovirus resistance protein A), RIG1 (Retinoic acid-inducible gene I), and MDA5 (Melanoma differentiation-associated protein 5), are involved in the recognition of pathogen-associated molecular patterns (PAMPs) and initiation of the innate immune response, including the production of interferons and other cytokines. Studying SNPs in these genes can provide insights into how genetic variations may influence individual susceptibility to infections, autoimmune diseases, and response to vaccines or immunotherapies. It is an exciting area of research with implications for personalized medicine and understanding the complexities of the immune system. In infants and toddlers, whose immune systems are still developing, these subtle genetic differences can have a more pronounced impact. When the immune response is not robust enough to completely clear the virus, it can lead to persistent infection, increasing the risk of SSPE development over time. Understanding these genetic predispositions is crucial for identifying individuals who may be at higher risk of developing SSPE following measles infection. This knowledge can inform targeted interventions and potentially help mitigate the risk of this devastating complication [39,40,41,42,43,44,45,46,47,48]. Indeed, the interplay between genetic variations and immune responses is crucial in understanding susceptibility to diseases like SSPE (subacute sclerosing panencephalitis), a rare but severe complication of measles virus infection. SSPE arises when the measles virus persists in the brain, leading to progressive neurological deterioration. Genetic variations impacting immune responses can influence how the body reacts to the measles virus. For instance, certain genetic factors may affect how efficiently the immune system clears the virus or regulates its activity. Additionally, variations in genes involved in immune signalling pathways could contribute to an overly aggressive immune response, leading to tissue damage. However, while we recognize the importance of genetic predispositions, the precise mechanisms underlying SSPE development remain elusive. Research efforts are ongoing to unravel the intricate interactions between the virus, the immune system, and genetic factors. Understanding these mechanisms is crucial for developing targeted therapies and preventive strategies for SSPE and other viral-induced neurological complications.

Tau protein in pathogenesis of SSPE
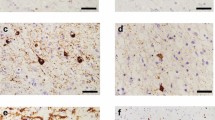
Anomalous forms of tau in the cerebrospinal fluid (CSF) of SSPE patients suggests a potential link between SSPE and tau pathology, possibly indicating neurodegenerative processes in the disease. This finding could have significant implications for understanding the mechanisms underlying SSPE and potentially for developing new diagnostic or therapeutic approaches [49]. Research based studies has indicated that the measles virus can persist in the central nervous system, affecting both oligodendrocytes (cells responsible for producing the myelin sheath that insulates nerve fibers) and neurons. This persistence of the virus in the brain is believed to play a role in the pathogenesis of SSPE [50]. Research investigating tauopathies in treated SSPE patients through brain autopsies is essential for understanding the underlying pathology of this condition. In these studies, researchers typically perform detailed neuropathological examinations of postmortem brain tissue from SSPE patients. They pay attention for evidence of tau protein accumulation, neuronal loss, gliosis (reactive changes in glial cells), and other pathological features associated with tauopathies. Such studies can provide insights into the mechanisms of disease progression, potential therapeutic targets, and diagnostic markers. This indicates that while the antiviral therapies may help in suppressing the virus, they may not be sufficient to prevent the development or progression of tauopathies in SSPE patients. This underscores the complexity of treating SSPE and the need for further research into more comprehensive treatment strategies [3]. NFTs are actually abnormal aggregates of tau protein within nerve cells. These tangles are formed due to the abnormal phosphorylation of tau protein, which causes it to become tangled and disrupt the normal functioning of the nerve cells. Such abnormal event was also observed in the SSPE patient’s brain [51, 52]. Such types of NFTs were excessively observed in the outermost layer of the limbic cortex, whereas globose-type NFTs occupied the positioning in the brainstem tegmentum. Entire of all such types of NFTs were resembled with the features of 3R + 4R tauopathies. While both SSPE and CTE involve tauopathies, their underlying causes are different. SSPE is caused by persistent infection with the measles virus, leading to brain inflammation and tau pathology, whereas CTE is associated with repeated head trauma. However, the similarities in the progression and presentation of tauopathies in these conditions may warrant further investigation into potential overlapping mechanisms or pathways involved in their development [3]. The relationship between tau pathology and the measles virus in SSPE is complex and not fully understood. However, studies have shown that the measles virus can directly or indirectly lead to the abnormal phosphorylation (a type of modification) of tau protein, which may contribute to the formation of NFTs. Additionally, the presence of viral intranuclear inclusions, which are characteristic of measles virus infection, has been observed in SSPE-affected brains [53]. Phosphorylated tau was dispersed throughout the entire CNS of the patient. However, there was a decrease in the quantity of phosphorylated tau in specific regions, such as the medial temporal lobe, cerebellum, and medulla oblongata. The conclusion that tauopathies and SSPE might be the result of diffuse brain inflammation generated through the measles virus suggests a potential indirect mechanism by which the virus could contribute to these conditions. This could imply that the measles virus triggers an inflammatory response in the brain, which then leads to the development of tauopathies and SSPE. However, further research would be needed to fully understand these mechanisms and their implications for treatment and prevention [3]. Indeed, the exact cause of NFT formation in SSPE patients is not fully understood, but researchers have proposed various hypotheses. One hypothesis suggests that viral infection leads to disturbances in cellular metabolism, affecting enzymes responsible for modifying tau proteins, which are crucial for maintaining the structure and function of neurons. Tau proteins normally undergo modifications, such as phosphorylation and dephosphorylation, regulated by enzymes like proteases, kinases, and phosphatases. Disruption in these processes could potentially result in the abnormal accumulation and aggregation of tau proteins into neurofibrillary tangles, contributing to the neurodegenerative pathology seen in SSPE (Fig. 5) [54]. Tissue culture-based studies were performed and neuroblastoma cell lines were infected with the measles virus. The outcomes of these studies were indicate that persistent infection leads to an increase in the activity of protein kinase C, which in turn is directly associated with enhanced viral gene expression. These findings suggest that when cells are infected with the measles virus, it alters cellular protein metabolism, particularly those related to the functional activity of neuronal cells. This could have implications for understanding how the virus interacts with and affects neural cells, potentially shedding light on the mechanisms underlying measles infection in the nervous system [55]. Production of NFTs in SSPE is powerfully connected to the infection of measles virus and determined at the level of the cell. NFTs configurations have not well established. It is possible that the connection of NFTs arrangement in patient with SSPE may be responsible for the existence of the nucleic acids of the measles virus as well as the immunoreactivity of tau. Immunoreactivity of tau protein is demonstrated a distinct connection to the period of disease. Positivity in the immunoreactivity was observed in those SSPE patients, which survival was 1 year duration and highest positivity was observed in the patient with longer survival. Furthermore, there was a strong connection observed in between the measles virus and tau protein in each distinct cell. This fact was observed in the majority of rigorously affected SSPE patient [52]. NFTs and tau have been revealed in the brain of 20% of patient with SSPE. It was found that reduce quantity of p-tau and no observation of meticulous alteration in S100-B and t-tau. NFT is not along with the pathogenesis procedures in SSPE, at least in children. On the other hand, the continuing intracellular infection of virus may be altering the functional role of kinases, which are responsible for phosphorylating the tau protein. Nevertheless, it appears probable that tau polymerization happening in SSPE may contribute to their neurological characteristics. Staging of SSPE is fundamentally grounded on the symptoms of myoclonus and motor. NFTs deterioration has possibly minute involvement to these characteristics, whereas signs of cortical dysfunction like as retreated language capability and distressed surroundings rhythm on EEG were linked with elevated tau levels, signifying a closer association between these roles and NFTs [49].

Interruptions in the IGF-1 and insulin pathways may play a role in the phosphorylation of tau protein and neurodegeneration seen in SSPE. Elevated levels of IGF-1 were found in the CSF and serum of SSPE patients in the fourth stage of the disease. This suggests a potential role of IGF-1 in the progression of the disease. Enhanced IGF-1 levels might hinder the phosphorylation-based modification of tau protein. This interference could have implications for the development of NFTs, a hallmark of neurodegenerative diseases. Microglia is identified as the primary source of IGF-1 in the indignant brain. This suggests a potential link between neuro-inflammation and the regulation of IGF-1 in the context of SSPE. These points suggest a complex interplay between IGF-1, tau protein phosphorylation, and neurodegeneration in SSPE [56]. In the fourth stage of SSPE, which is the most advanced stage, there is significant neuronal loss and the formation of microglial nodules in the brain. These microglial nodules represent areas of activated microglial cells, which are the resident immune cells of the central nervous system. They become activated in response to neuronal injury or infection [57, 58]. Enhanced IGF-1 level is associated to reactive gliosis and is more probable to be secondary to decreased IGF-1 signalling due to the loss of IGF-1 receptors alongside with neurons [59,60,61,62,63].
Prognostic significance of tau protein in SSPE
The prognostic significance of tau protein in subacute sclerosing panencephalitis (SSPE) is not well-established, and tau protein is not typically used as a specific prognostic marker in the context of SSPE. SSPE is a rare, progressive, and typically fatal neurological disorder caused by persistent infection with the measles virus (MeV) [64]. The prognosis in SSPE is primarily determined by various clinical and neurological factors, including the stage of the disease, age at onset, and the extent of neurological involvement [65]. While tau protein is not typically considered a prognostic marker in SSPE, it is important to note that SSPE primarily involves the accumulation of measles virus-infected cells in the brain, leading to a progressive and devastating neurodegenerative condition. The primary pathogenic mechanisms in SSPE are related to the persistent presence of the measles virus, the immune response to it, and the associated neuronal damage [66]. The prognosis for SSPE is generally poor, with the disease often leading to severe disability and eventually death. Given the complexity of SSPE and the lack of specific biomarkers for prognosis, clinical assessment and monitoring of neurological and cognitive function are critical in determining the course of the disease and guiding treatment decisions.
Therapeutic approaches of SSPE
Because there is no cure for SSPE, treatment options aim to alleviate symptoms, slow disease progression, and improve quality of life. There are several treatment options that have been used among patients with SSPE. Antiviral medications such as interferon-alpha and ribavirin have been used in some cases to try to suppress the replication of the measles virus. However, their effectiveness in SSPE remains uncertain. Drugs that modulate the immune system, such as intravenous immunoglobulin (IVIG) and corticosteroids have been used to try to reduce inflammation and immune-mediated damage in the brain [11, 67,68,69]. Medications may be prescribed to manage symptoms such as seizures, myoclonus and involuntary movements. These may include anti seizure medications , muscle relaxants, and antipsychotics. Patients with SSPE often require extensive supportive care to address their physical and cognitive disabilities. This may involve physical therapy, occupational therapy, speech therapy, and psychological support. In some cases, experimental treatments or therapies aimed at boosting the immune response or targeting specific aspects of the disease process may be considered [11, 67,68,69,70]. However, these treatments are not standardized and are typically only available through clinical trials or under compassionately used protocols. It is important to note that the effectiveness of these treatments can vary widely among individuals, and there is currently no consensus on the optimal treatment approach for SSPE. Management is typically tailored to the individual patient's symptoms, disease progression, and response to treatment [11, 70]. Using a recombinant adenovirus, expressing small interfering RNA to inhibit replication of the measles virus and SSPE virus is a promising approach. It seems like Otaki et al. demonstrated its efficacy convincingly in their study. This method could potentially offer a targeted and efficient therapeutic strategy against SSPE, addressing a critical need in managing this condition [70]. Trihexyphenidyl, clonazepam and valproate are often used to manage the myoclonus jerks and involuntary movements in patients. But, these medications are only symptomatic and are not without side effects, hence treatment needs to be individualized. Plasma exchange therapy has also been tried, but without any short-term or long-term beneficial effects [66]. It is interesting that the mechanism of action of stem cell transplantation is thought to involve the release of anti-apoptotic (anti-cell death) and anti-inflammatory mediators, although the exact mechanisms are not fully understood. However, despite this potential mechanism, there is no evidence of benefit from such therapy [67]. In SSPE, where inflammation in the brain plays a significant role, the anti-inflammatory properties of the ketogenic diet could be potentially explored to help mitigate some of the neurological symptoms. Additionally, its metabolic effects, such as promoting ketosis, might provide an alternative energy source for neurons, possibly offsetting some of the neuronal damage caused by the disease process. Cognitive improvement with cessation of myoclonic jerks stopped has aptly been demonstrated in single case reports [69].
Discussion
The review attempts to discuss the importance of tau protein in neuronal function and its involvement in various neurodegenerative diseases. The pathogenesis of SSPE is primarily focussed around the tau protein, tauopathies and certain pivotal genes; all of which are potential therapeutic targets. This would lead to development of effective and efficient treatment strategies thereby improving quality of life in patients. While the primary role of tau was initially believed to be in stabilizing microtubules in neurons, several research based studies emphasize additional and yet unexplored functions. The non-filamentous form of hyperphosphorylated tau potentially being held responsible for early behavioural deficits is rather intriguing. It suggests that tau pathology might manifest differently than the classical view of tau aggregation into neurofibrillary tangles. Understanding the role of phosphorylated tau in development and signal transduction could indeed shed light on pathways that are dysregulated in the neurodegenerative diseases. By exploring these alternative functions of tau, researchers might unravel novel therapeutic targets and develop new strategies for combating neurodegenerative disorders. This underscores the importance of continued investigation into tau biology and its implications for human health [1, 71].
Development of tauopathy following subacute sclerosing panencephalitis (SSPE) is likely due to widespread brain inflammation caused by the measles virus rather than direct viral action itself. This inference is drawn from the observation that the distribution patterns of phosphorylated tau and measles virus are independent of each other. Furthermore, existing antiviral treatments for SSPE focus on suppressing the measles virus but are unable to effectively halt the progression of secondary tauopathy. To address this, a combination therapy approach involving both antiviral drugs and treatments targeting tau proteins could offer promising outcomes for SSPE patients in the future. This combined approach may better address the dual mechanisms underlying the disease and improve patient prognosis [3, 11, 68,69,70].
In SSPE, the presence of abundant filamentous inclusions made of various brain tau isoforms resembles what is seen in AD and CTE. While it is known that inflammation and microglial cell activation can influence tau assembly, the exact mechanisms remain unclear. Studies in mice have suggested that microglial activation can promote tau assembly, which is significant because similar activation is observed in neurodegenerative diseases beyond SSPE and CTE, with AD being the most studied among them. Understanding the intricate links between neuro-inflammation and tau assembly could offer valuable insights into the pathogenesis of these diseases and potentially lead to better therapeutic approaches. However, further research is needed to elucidate these connections[9, 71].
The precise mechanisms underlying SSPE are not fully understood, but several factors contribute to its development. One key factor is thought to be mutations in the measles virus genome, particularly in genes encoding the M, F, and H proteins. These mutations may lead to alterations in viral replication, assembly, and neuro-virulence. These biased hyper-mutations observed in the viral genome, especially in the M gene, may impair the virus's ability to produce infectious viral particles while enhancing its neuro-tropism and persistence in the central nervous system. The consequential mutations in the M, F, and H proteins likely play a role in the pathogenesis of SSPE by altering the interactions between the virus and host cells, leading to neuronal damage and inflammation. While SSPE remains a rare complication of measles infection, understanding the molecular mechanisms underlying its development is crucial for developing targeted therapies and preventive strategies [34, 72].
This hypothesis presents an intriguing connection between the measles virus and neurodegenerative conditions like tauopathies and SSPE. The idea that the virus could induce brain inflammation, subsequently leading to the development of these conditions, highlights the intricate relationship between viral infections and neurological health. Understanding the mechanisms underlying these associations could pave the way for innovative treatment and prevention strategies. If further research confirms this link, it might suggest avenues for targeting inflammation in the brain as a means of mitigating the development or progression of tauopathies and SSPE in individuals affected by measles virus infection. It is also essential to explore how vaccination and other preventive measures against measles might impact the incidence of these neurological conditions. Preventing measles infections could potentially reduce the risk of associated neurological complications, providing yet another incentive for vaccination efforts [3, 50,51,52,53,54,55].
There are significant challenges in treating SSPE (subacute sclerosing panencephalitis), particularly in terms of disease modification. The lag between clinical descriptions and effective treatment strategies underscores the complexity of the condition. It seems crucial for future research to focus on newer approaches, especially targeting the early inflammatory stages of SSPE, before irreversible neurodegeneration occurs. The lack of large-scale randomized trials and limited patient numbers in studies make it difficult to assess the efficacy of current therapeutic strategies accurately. This underscores the need for more robust research methodologies and collaborations to gather meaningful data. Additionally, the potential use of ketogenic diet strategies in SSPE treatment is challenging , and requires careful consideration due to its demanding nature and the need for close monitoring. This highlights the importance of personalized approaches and thorough patient evaluation, especially given the overlapping clinical presentation of SSPE with conditions, like autoimmune encephalitis. Addressing the gaps in understanding the pathogenesis of SSPE is also crucial for developing more effective treatment strategies. Collaborative efforts among researchers, clinicians, and patients will be essential in advancing our understanding and management of this challenging condition [11, 67,68,69,70,71,72].
Conclusion
Indeed, the role of tauopathies in subacute sclerosing panencephalitis (SSPE) is a crucial aspect of the disease pathology. Tau proteins are essential for the normal functioning of neurons, but their abnormal accumulation, forming neurofibrillary tangles, is a hallmark of several neurodegenerative diseases, including SSPE. Understanding the molecular mechanisms behind tau accumulation in SSPE is vital for several reasons. Firstly, it can shed light on the specific pathways through which the measles virus, (the causative agent of SSPE), triggers neurodegeneration. Secondly, it can help identify potential therapeutic targets aimed at preventing or slowing down tau aggregation. Additionally, understanding the clinical implications of tauopathy in SSPE can aid in the development of diagnostic tools to detect the disease at earlier stages and monitor its progression effectively. Given the complexity of SSPE and the multifaceted interactions between viral infection, neuro-inflammation, and tau pathology, further research is undoubtedly warranted. By elucidating the precise links between these factors, researchers can pave the way for the development of more targeted and effective treatments for SSPE, ultimately improving the prognosis for affected patients. It is a challenging task, but one that holds immense promise for the future.
Data availability
Not applicable
Abbreviations
- SSPE:
-
Subacute sclerosing panencephalitis
- PSP:
-
Progressive supranuclear palsy
- CTE:
-
Chronic traumatic encephalopathy
- PART:
-
Primary age-related tauopathy
References
Lee G, Leugers CJ. Tau and tauopathies. Prog Mol Biol Transl Sci. 2012;107:263. https://doi.org/10.1016/B978-0-12-385883-2.00004-7.
Zhang Y, Wu KM, Yang L, Dong Q, Yu JT. Tauopathies: new perspectives and challenges. Mol Neurodegener. 2022;17:1–29. https://doi.org/10.1186/S13024-022-00533-Z.
Miyahara H, Akagi A, Riku Y, Sone J, Otsuka Y, Sakai M, et al. Independent distribution between tauopathy secondary to subacute sclerotic panencephalitis and measles virus: an immunohistochemical analysis in autopsy cases including cases treated with aggressive antiviral therapies. Brain Pathol. 2022;32: e13069. https://doi.org/10.1111/BPA.13069.
Barbier P, Zejneli O, Martinho M, Lasorsa A, Belle V, Smet-Nocca C, et al. Role of tau as a microtubule-associated protein: structural and functional aspects. Front Aging Neurosci. 2019. https://doi.org/10.3389/FNAGI.2019.00204.
Catarina Silva M, Haggarty SJ. Tauopathies: deciphering disease mechanisms to develop effective therapies. Int J Mol Sci. 2020;21:8948. https://doi.org/10.3390/IJMS21238948.
Bellucci A, Bugiani O, Ghetti B, Spillantini MG. Presence of reactive microglia and neuroinflammatory mediators in a case of frontotemporal dementia with P301S mutation. Neurodegener Dis. 2011;8(4):221–9. https://doi.org/10.1159/000322228.
Upadhyayula PS, Yang J, Yue JK, Ciacci JD. Subacute sclerosing panencephalitis of the brainstem as a clinical entity. Med Sci. 2017;5:26. https://doi.org/10.3390/MEDSCI5040026.
Holmes BB, Conell-Price J, Kreple CJ, Ashraf D, Betjemann J, Rosendale N. Adult-onset subacute sclerosing panencephalitis with a 30-year latent period. Neurohospitalist. 2020;10:127. https://doi.org/10.1177/1941874419869713.
Qi C, Hasegawa M, Takao M, Sakai M, Sasaki M, Mizutani M, et al. Identical tau filaments in subacute sclerosing panencephalitis and chronic traumatic encephalopathy. Acta Neuropathol Commun. 2023;11:74. https://doi.org/10.1186/s40478-023-01565-2.
Gong C-X, Iqbal K. Hyperphosphorylation of microtubule-associated protein tau: a promising therapeutic target for Alzheimer disease. Curr Med Chem. 2008;15:2321. https://doi.org/10.2174/092986708785909111.
Mekki M, Eley B, Hardie D, Wilmshurst JM. Subacute sclerosing panencephalitis: clinical phenotype, epidemiology, and preventive interventions. Dev Med Child Neurol. 2019;61:1139–44. https://doi.org/10.1111/DMCN.14166.
Saha V, Jacob John T, Mukundan P, Gnanamuthu C, Prabhakar S, Arjundas G, et al. High incidence of subacute sclerosing panencephalitis in south India. Epidemiol Infect. 1990;104:151–6. https://doi.org/10.1017/S0950268800054637.
Garg RK, Mahadevan A, Malhotra HS, Rizvi I, Kumar N, Uniyal R. Subacute sclerosing panencephalitis. Rev Med Virol. 2019;29: e2058. https://doi.org/10.1002/RMV.2058.
Hashimoto K, Hosoya M. Advances in Antiviral Therapy for Subacute Sclerosing Panencephalitis. Molecules. 2021;26:427. https://doi.org/10.3390/MOLECULES26020427.
Jafri SK, Kumar R, Ibrahim S. Subacute sclerosing panencephalitis – current perspectives. Pediatric Health Med Ther. 2018;9:67. https://doi.org/10.2147/PHMT.S126293.
Saurabh K, Singh V, Pathak A, Chaurasia R. Subacute sclerosing pan encephalitis: an update. J Clin Sc Res. 2021;10:35. https://doi.org/10.4103/JCSR.JCSR_68_20.
Ludwig PE, Reddy V, Varacallo M. Neuroanatomy, Neurons. StatPearls 2023.
Brunello CA, Merezhko M, Uronen RL, Huttunen HJ. Mechanisms of secretion and spreading of pathological tau protein. Cell Mol Life Sci. 2020;77:1721–44. https://doi.org/10.1007/S00018-019-03349-1.
Miller JH, Das V. Potential for treatment of neurodegenerative diseases with natural products or synthetic compounds that stabilize microtubules. Curr Pharm Des. 2020;26:4362–72. https://doi.org/10.2174/1381612826666200621171302.
Gao Y-L, Wang N, Sun F-R, Cao X-P, Zhang W, Yu J-T. Tau in neurodegenerative disease. Ann Transl Med. 2018;6:175–175. https://doi.org/10.21037/ATM.2018.04.23.
Metaxas A, Kempf SJ. Neurofibrillary tangles in Alzheimer’s disease: elucidation of the molecular mechanism by immunohistochemistry and tau protein phospho-proteomics. Neural Regen Res. 2016;11:1579. https://doi.org/10.4103/1673-5374.193234.
Gendron TF. The role of tau in neurodegeneration. Mol Neurodegener. 2009;4:1–19. https://doi.org/10.1186/1750-1326-4-13/FIGURES/1.
Rawat P, Sehar U, Bisht J, Selman A, Culberson J, Reddy PH. Phosphorylated Tau in Alzheimer’s disease and other tauopathies. Int J Mol Sci. 2022;23:12841. https://doi.org/10.3390/IJMS232112841.
Duyckaerts C, Clavaguera F, Potier MC. The prion-like propagation hypothesis in Alzheimer’s and Parkinson’s disease. Curr OpinNeurol. 2019;32:266–71. https://doi.org/10.1097/WCO.0000000000000672.
Almansoub HAMM, Tang H, Wu Y, Wang DQ, Mahaman YAR, Wei N, et al. Tau abnormalities and the potential therapy in Alzheimer’s disease. J Alzheimers Dis. 2019;67:13–33. https://doi.org/10.3233/JAD-180868.
Alquezar C, Arya S, Kao AW. Tau Post-translational modifications: dynamic transformers of tau function, degradation, and aggregation. Front Neurol. 2021. https://doi.org/10.3389/FNEUR.2020.595532.
Ercan-Herbst E, Ehrig J, Schöndorf DC, Behrendt A, Klaus B, Gomez Ramos B, et al. A post-translational modification signature defines changes in soluble tau correlating with oligomerization in early stage Alzheimer’s disease brain. Acta Neuropathol Commun. 2019. https://doi.org/10.1186/S40478-019-0823-2.
Kametani F, Yoshida M, Matsubara T, Murayama S, Saito Y, Kawakami I, et al. Comparison of common and disease-specific post-translational modifications of pathological tau associated with a wide range of tauopathies. Front Neurosci. 2020. https://doi.org/10.3389/FNINS.2020.581936.
Miao J, Shi R, Li L, Chen F, Zhou Y, Tung YC, et al. Pathological Tau from Alzheimer’s brain induces site-specific hyperphosphorylation and sds- and reducing agent-resistant aggregation of tau in vivo. Front Aging Neurosci. 2019. https://doi.org/10.3389/FNAGI.2019.00034.
Polanco JC, Götz J. Exosomal and vesicle-free tau seeds—propagation and convergence in endolysosomal permeabilization. FEBS J. 2022;289:6891–907. https://doi.org/10.1111/FEBS.16055.
Zhang H, Cao Y, Ma L, Wei Y, Li H. Possible mechanisms of tau spread and toxicity in Alzheimer’s disease. Front Cell Dev Biol. 2021. https://doi.org/10.3389/FCELL.2021.707268.
Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci. 2015;18:1584–93. https://doi.org/10.1038/nn.4132.
Guan YH, Zhang LJ, Wang SY, Deng YD, Zhou HS, Chen DQ, et al. The role of microglia in Alzheimer’s disease and progress of treatment. Ibrain. 2022;8:37–47. https://doi.org/10.1002/IBRA.12023.
Samia P, Oyieke K, Tunje D, Udwadia-Hegde A, Feemster K, Oncel I, et al. Options in the treatment of subacute sclerosing panencephalitis: implications for low resource areas. Curr Treat Options Neurol. 2022;24:99. https://doi.org/10.1007/S11940-022-00710-X.
Bhattacharjee S, Yadava PK. Measles virus: background and oncolytic virotherapy. BiochemBiophys Rep. 2018;13:58. https://doi.org/10.1016/J.BBREP.2017.12.004.
Honda T, Yoneda M, Sato H, Kai C, Honda T, Yoneda M, et al. Pathogenesis of encephalitis caused by persistent measles virus infection. Encephalitis. 2013. https://doi.org/10.5772/54434.
Ono N, Tatsuo H, Hidaka Y, Aoki T, Minagawa H, Yanagi Y. Measles viruses on throat swabs from measles patients use signaling lymphocytic activation molecule (CDw150) but not CD46 as a cellular receptor. J Virol. 2022;75:4399–401. https://doi.org/10.1128/JVI.75.9.4399-4401.2001.
Angius F, Smuts H, Rybkina K, Stelitano D, Eley B, Wilmshurst J, et al. Analysis of a subacute sclerosing panencephalitis genotype B3 virus from the 2009–2010 South African measles epidemic shows that hyperfusogenic F proteins contribute to measles virus infection in the brain. J Virol. 2019. https://doi.org/10.1128/JVI.01700-18.
Torisu H, Kusuhara K, Kira R, Bassuny WM, Sakai Y, Sanefuji M, et al. Functional MxA promoter polymorphism associated with subacute sclerosing panencephalitis. Neurology. 2004;62:457–60. https://doi.org/10.1212/01.WNL.0000106940.95749.8E.
Ishizaki Y, Takemoto M, Kira R, Kusuhara K, Torisu H, Sakai Y, et al. Association of toll-like receptor 3 gene polymorphism with subacute sclerosing panencephalitis. J Neurovirol. 2008;14:486–91. https://doi.org/10.1080/13550280802298120.
Karakas-Celik S, Piskin IE, Keni MF, Calik M, Iscan A, Dursun A. May TLR4 Asp299Gly and IL17 His161Arg polymorphism be associated with progression of primary measles infection to subacute sclerosing panencephalitis? Gene. 2014;547:186–90. https://doi.org/10.1016/J.GENE.2014.03.056.
Dundar NO, Gencpinar P, Sallakci N, Duman O, Haspolat S, Anlar B, et al. Interleukin-12 (-1188) A/C and interferon-γ (+874) A/T gene polymorphisms in subacute sclerosing panencephalitis patients. J Neurovirol. 2016;22:661–5. https://doi.org/10.1007/S13365-016-0442-7.
Yilmaz V, Demirbilek V, Gürses C, Yentür S, Uysal S, Yapici Z, et al. Interleukin (IL)-12, IL-2, interferon-gamma gene polymorphisms in subacute sclerosing panencephalitis patients. J Neurovirol. 2007;13:410–5. https://doi.org/10.1080/13550280701455383.
Inoue T, Kira R, Nakao F, Ihara K, Bassuny WM, Kusuhara K, et al. Contribution of the interleukin 4 gene to susceptibility to subacute sclerosing panencephalitis. Arch Neurol. 2002;59:822–7. https://doi.org/10.1001/ARCHNEUR.59.5.822.
Piskin IE, Karakas-Celik S, Calik M, Abuhandan M, Kolsal E, Genc GC, et al. Association of interleukin 18, interleukin 2, and tumor necrosis factor polymorphisms with subacute sclerosing panencephalitis. DNA Cell Biol. 2013;32:336–40. https://doi.org/10.1089/DNA.2013.1997.
Yentur SP, Aydin HN, Gurses C, Demirbilek V, Kuru U, Uysal S, et al. Granzyme B gene polymorphism associated with subacute sclerosing panencephalitis. Neuropediatrics. 2014;45:309–13. https://doi.org/10.1055/S-0034-1378129.
Piskin I, Calk M, Abuhandan M, Kolsal E, Celik S, Iscan A. PD-1 gene polymorphism in children with subacute sclerosing panencephalitis. Neuropediatrics. 2013;44:187–90. https://doi.org/10.1055/S-0033-1338134.
Uygun DFK, Uygun V, Burgucu D, Ekinci NÇ, Sallakçı N, Filiz S, et al. Role of the Th1 and Th17 pathway in subacute sclerosing panencephalitis. J Child Neurol. 2019;34:815–9. https://doi.org/10.1177/0883073819860631.
Yuksel D, Yilmaz D, Uyar NY, Senbil N, Gurer Y, Anlar B. Tau proteins in the cerebrospinal fluid of patients with subacute sclerosing panencephalitis. Brain Dev. 2010;32:467–71. https://doi.org/10.1016/J.BRAINDEV.2009.11.009.
Isaacson SH, Asher DM, Godec MS, Gibbs CJ, Gajdusek DC. Widespread, restricted low-level measles virus infection of brain in a case of subacute sclerosing panencephalitis. Acta Neuropathol. 1996;91:135–9. https://doi.org/10.1007/S004010050404.
Maderna E, Fugnanesi V, Morbin M, Cacciatore F, Spinello S, Godani M, et al. Measles inclusion-body encephalitis: neuronal phosphorylated tau protein is present in the biopsy but not in the autoptic specimens of the same patient. Brain Pathol. 2016;26:542–6. https://doi.org/10.1111/BPA.12332.
McQuaid S, Allen IV, McMahon J, Kirk J. Association of measles virus with neurofibrillary tangles in subacute sclerosing panencephalitis: a combined in situ hybridization and immunocytochemical investigation. Neuropathol Appl Neurobiol. 1994;20:103–10. https://doi.org/10.1111/J.1365-2990.1994.TB01168.X.
Bancher C, Leitner H, Jellinger K, Eder H, Setinek U, Fischer P, et al. On the relationship between measles virus and Alzheimer neurofibrillary tangles in subacute sclerosing panencephalitis. Neurobiol Aging. 1996;17:527–33. https://doi.org/10.1016/0197-4580(96)00069-3.
Trojanowski JQ, Schuck T, Schmidt ML, Lee VMY. Distribution of tau proteins in the normal human central and peripheral nervous system. J Histochem Cytochem. 1989;37:209–15. https://doi.org/10.1177/37.2.2492045.
Lu Q, Wood JG. Functional studies of Alzheimer’s disease tau protein. J Neurosci. 1993;13:508–15. https://doi.org/10.1523/JNEUROSCI.13-02-00508.1993.
Mashayekhi F, Mirzajani E, Naji M, Azari M. Expression of insulin-like growth factor-1 and insulin-like growth factor binding proteins in the serum and cerebrospinal fluid of patients with Parkinson’s disease. J Clin Neurosci. 2010;17:623–7. https://doi.org/10.1016/J.JOCN.2009.08.013.
Yüksel D, Diren B, Ulubay H, Altunbaşak Ş, Anlar B. Neuronal loss is an early component of subacute sclerosing panencephalitis. Neurology. 2014;83:938–44. https://doi.org/10.1212/WNL.0000000000000749.
Anlar B, Söylemezoğlu F, Aysun S, Köse G, Belen D, Yalaz K. Tissue inflammatory response in subacute sclerosing panencephalitis (SSPE). J Child Neurol. 2001;16:895–900. https://doi.org/10.1177/088307380101601206.
Chesik D, Wilczak N, De Keyser J. The insulin-like growth factor system in multiple sclerosis. Int Rev Neurobiol. 2007;79:203–26. https://doi.org/10.1016/S0074-7742(07)79009-8.
Johansson P, Åberg D, Johansson JO, Mattsson N, Hansson O, Ahrén B, et al. Serum but not cerebrospinal fluid levels of insulin-like growth factor-I (IGF-I) and IGF-binding protein-3 (IGFBP-3) are increased in Alzheimer’s disease. Psychoneuroendocrinology. 2013;38:1729–37. https://doi.org/10.1016/J.PSYNEUEN.2013.02.006.
Carro E, Torres-Aleman I. The role of insulin and insulin-like growth factor I in the molecular and cellular mechanisms underlying the pathology of Alzheimer’s disease. Eur J Pharmacol. 2004;490:127–33. https://doi.org/10.1016/j.ejphar.2004.02.050.
Moloney AM, Griffin RJ, Timmons S, O’Connor R, Ravid R, O’Neill C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol Aging. 2010;31:224–43. https://doi.org/10.1016/J.NEUROBIOLAGING.2008.04.002.
Yilmaz D, Yüksel D, Gökkurt D, Oguz H, Anlar B. Increased insulin-like growth factor-1 levels in cerebrospinal fluid of advanced subacute sclerosing panencephalitis patients. Eur J Paediatr Neurol. 2016;20:611–5. https://doi.org/10.1016/J.EJPN.2016.03.007.
Papetti L, Amodeo ME, Sabatini L, Baggieri M, Capuano A, Graziola F, et al. Subacute sclerosing panencephalitis in children: the archetype of non-vaccination. Viruses. 2022. https://doi.org/10.3390/V14040733.
Gutierrez J, Issacson RS, Koppel BS. Subacute sclerosing panencephalitis: an update. Dev Med Child Neurol. 2010;52:901–7. https://doi.org/10.1111/J.1469-8749.2010.03717.X.
Lizarraga KJ, Gutierrez J, Singer C. Subacute Sclerosing Panencephalitis. The Curated Reference Collection in Neuroscience and Biobehavioral Psychology 2023:187–9. https://doi.org/10.1016/B978-0-12-809324-5.00824-5.
Garg RK. Subacute sclerosing panencephalitis. J Neurol. 2008;255(12):1861–71. https://doi.org/10.1007/s00415-008-0032-6.
Garg M, Arora A, Kulkarni SD, Hegde AU, Shah KN. Subacute sclerosing panencephalitis (SSPE): experience from a tertiary-care pediatric center. J Neurosci Rural Pract. 2022;13(2):315–20. https://doi.org/10.1055/s-0041-1740612.
Valente M, Del Negro I, Bagatto D, et al. Clinical and magnetic resonance study of a case of subacute sclerosing panencephalitis treated with ketogenic diet. BMJ Neurol Open. 2021;3(2): e000176. https://doi.org/10.1136/bmjno-2021-000176.
Oyama F, Kotliarova S, Harada A, et al. Gem GTPase and tau: morphological changes induced by gem GTPase in cho cells are antagonized by tau. J Biol Chem. 2004;279(26):27272–7. https://doi.org/10.1074/jbc.M401634200.
Maphis N, Xu G, Kokiko-Cochran ON, et al. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain. 2015;138(Pt 6):1738–55. https://doi.org/10.1093/brain/awv081.
Hotta H, Jiang DP, Nagano-Fujii M. SSPE virus and pathogenesis. Nihon Rinsho. 2007;65(8):1475–80.
Author information
Authors and Affiliations
Contributions
NP and NKS performed the conceptualization, design, literature search review, and writing the original draft. AK and IH edited the drafts. DJ performed the final review, supervision and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare that there are no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pandey, N., Srivastava, N.K., Kumar, A. et al. A comprehensive expedition of tauopathies in subacute sclerosing panencephalitis (SSPE): a narrative review. Egypt J Neurol Psychiatry Neurosurg 60, 96 (2024). https://doi.org/10.1186/s41983-024-00860-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41983-024-00860-6