
Abstract
Background
CADASIL is the most common single-gene disorder causing ischemic stroke. CADASIL has been linked to mutations in NOTCH3 gene, due to heterozygous missense mutations. The disease is of insidious onset, presenting with initial clinical features in third and fourth decade of life. However, it is now being increasingly acknowledged that individual clinical presentation, age, time of onset as well as disease severity are quite varied among patients with CADASIL most likely leading to under- or mis-diagnosis. The authors thereby report a genetically confirmed case of CADASIL with atypical clinical course and findings.
Case presentation
A 48-year-old woman presented with complaints of episodic headache, relapsing–remitting neurological illness, progressive cognitive impairment, and acute-onset loss of speech and ambulation. She was earlier being treated as a case of CNS demyelination for 10 years. On examination, vital parameters were within normal limits. Neurological examination revealed that the patient was drowsy, not verbalizing, not obeying commands, with movement of all four limbs on painful stimuli, hypertonia of all limbs, grade 3 + deep tendon reflexes, bilateral striatal toe and extensor plantar response. Magnetic resonance imaging of brain showed involvement of anterior temporal lobe and external capsule along with multiple acute infarcts. Cerebrospinal fluid analysis was found to be normal. Exome sequencing revealed heterozygous missense mutation in exon 2 of NOTCH3 gene. A definite diagnosis of CADASIL was made and patient was started on fluoxetine and aspirin, following which there was significant improvement over 4–6 weeks. Patient is able to carry out daily activities independently although continues to have mild persistent cognitive impairment with excessive talking and over familiarity.
Conclusions
As CADASIL has a relapsing and partially remitting course with frequently observed varied clinical presentation, patients may receive treatment for demyelination which may not be necessary. Hence, detailed family history along with knowledge of characteristic magnetic resonance imaging findings seen in CADASIL can help discern the diagnosis.
Similar content being viewed by others

Introduction
Cerebral small-vessel disease is a condition that is frequently seen in association with ischemic stroke and dementia [1]. The abbreviation CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy) was first introduced in the year 1993 and the disease’s gene locus was additionally mapped to chromosome 19q12 [2]. Heterozygous missense mutations in NOTCH3 gene are linked to CADASIL [3, 4]. Most mutations involve exon 4 followed by 3, 6, 2 and 5 [5]. The disease is of insidious onset, presenting with initial clinical features in the third and fourth decade of life. Predominant clinical manifestations include migraine with aura, recurrent ischemic strokes, transient ischemic attacks, cognitive impairment, multiple affective symptoms such as depression, apathy, behavioral and mood changes [6,7,8]. However, it is now being recognized to a large extent that individual clinical presentation, age, and time of onset as well as disease severity are quite varied, even among individuals in the same family [9], most likely leading to under or misdiagnosis. As only a few cases of the disease have been reported from around the world especially from India, we report a genetically confirmed case of CADASIL with atypical clinical course and findings.
Case presentation
A 48-year-old woman presented to the neurology department with complaints of episodic headache since 20 years, relapsing–remitting neurological illness since 10 years, slowly progressive cognitive impairment since 6 months and acute-onset loss of speech and ambulation since one week. The headaches were episodic, amnestic, hemi-cranial, associated with visual aura and occasionally also with drooping of one eyelid and double vision, lasting for about 4–5 h, subsiding with analgesics. History of relapsing–remitting neurological illness for 10 years, with the 1st episode being right hemiparesis and 2nd episode left hemiparesis within a span of 2 years, both of which improved within few weeks. The present admission was following a self-limiting fever episode, after which there was worsening of cognitive impairment which gradually progressed to akinetic mutism and drowsy state over a week along with dual incontinence. Patient was earlier treated in another institute as multiple sclerosis (MS) with pulse steroids and beta-Interferon for 3 years, following which patient was switched to mycophenolate mofetil and treated for 7 years due to financial constraints. During the 10 years of treatment, patient continued to have headaches. On examination, vital parameters were stable and within normal limits. Neurological examination revealed that the patient was drowsy, not verbalizing, not obeying commands, there was movement of all four limbs on painful stimuli, hypertonia of all limbs, grade 3 + deep tendon reflexes, bilateral striatal toe and extensor plantar response.
Family history revealed that patient’s mother had migraine with aura and several episodes of stroke with first episode at around 40 years, developed progressive cognitive impairment at about 60 years of age and was bed bound, died of aspiration pneumonia at 65 years of age. All of patient’s four siblings also suffer from migraine with aura (Fig. 1).

Pedigree chart of patient
The patient’s routine blood investigations, serum homocysteine levels and CSF (cerebrospinal fluid) analysis were normal. Oligoclonal bands were not detected, and ANA (antinuclear antibody) profile was negative. MRI (magnetic resonance imaging) brain and screening of whole spine was done which revealed diffusion restriction involving corpus callosum and bilateral periventricular white matter in DWI (diffusion-weighted imaging) (Fig. 2A and C) and ADC (apparent diffusion co-efficient) images (Fig. 2B and D). T1 post-contrast showed no contrast enhancement (Fig. 3A, B). Hyperintense signal change involving bilateral frontal subcortical, deep subcortical white matter, bilateral periventricular white matter and corpus callosum (Fig. 4A, B) as well as hyperintense signal changes in bilateral anterior temporal and external capsule was seen on FLAIR (fluid-attenuated inversion recovery) images (Fig. 5A, B). TOF (time-of-flight) angiography was found to be normal. Based on the imaging findings, CADASIL was suspected, and NOTCH3 gene analysis was requested. A heterozygous missense variation in exon 2 of the NOTCH3 gene that resulted in the amino acid substitution of tryptophan for cysteine at codon 49 was detected.

A and C Showing DWI with B and D showing ADC images of multiple foci of diffusion restriction involving genu and body of corpus callosum and bilateral periventricular white matter suggestive of acute lacunar infarcts

A and B Showing T1 post-contrast images with no contrast enhancement of the diffusion restricting foci

A and B Showing FLAIR image with multiple hyperintense signal changes involving subcortical white matter of bilateral frontal lobes, bilateral periventricular white matter, bilateral corona radiata and corpus callosum

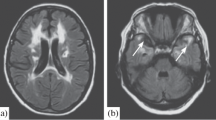
A and B Showing T2 FLAIR images with hyperintense signal changes in bilateral anterior temporal lobes and external capsule
At the end of history and examination three possible differentials were considered, namely relapsing–remitting multiple sclerosis (RRMS), CNS vasculitis and CADASIL. The present encephalopathy episode along with absence of lesions involving the spine and brainstem after 10 years of illness but with involvement of anterior temporal lobe were all considered to be unlikely of RRMS. In view of normal CSF and TOF angiogram findings with presence of positive family history, it was also less likely to be CNS vasculitis. Typical clinical and imaging features along with family history pointed towards the possibility of CADASIL. Hence, exome sequencing was requested which helped clinch the diagnosis.
The patient was given aspirin 75 mg/day for secondary stroke prevention. Apathy and mutism were treated with fluoxetine 20 mg/day and increased up to 40 mg/day. Patient showed significant improvement over 4–6 weeks. She currently is only on the above-mentioned medications. Patient has mild persistent cognitive impairment with excessive talking and over familiarity, however, has been able to carry out her daily activities independently. Patient continues to remain under follow-up and review.
Discussion
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is one of the broadly acknowledged heritable reason for stroke and vascular dementia in adults, with clinical and neuroimaging features bearing semblance to that of sporadic small-artery disease [10]. The clinical diagnosis of CADASIL is commonly based on the following conditions: (1) clinical onset at 40–50 years; (2) absence of stroke risk factors; (3) frequent lacunar infarction episodes leading to focal neurological deficits, cognitive impairment and dementia (migraine, cerebral infarction, and dementia in 30%, 85%, and 30–90% of patients, respectively); and (4) familial distribution of the disease or similar symptoms [11]. Patients with CADASIL have decreased life expectancy and there also currently exists no effective treatment for the disease.
Our 48-year-old patient presented with recurrent strokes, long-standing migraine with aura and encephalopathy. Historically and on clinical examination as our patient was in encephalopathy, MRI of the brain and spine was done to help with the diagnosis, however imaging findings with involvement of anterior temporal lobe and external capsule led us to suspect CADASIL rather than a case of CNS demyelination [12]. Recent studies on CADASIL have recognized the anterior temporal lobe and external capsules as sites of predilection for WMHs (white matter hyperintensities) which, if affected [13, 14], are very useful in differentiating it from other forms of small-vessel disease [14, 15]. According to Markus et al., anterior temporal lobe involvement has a much higher specificity than external capsule involvement [7], specificity being 100% in juxtapose to 45%, respectively. WMH lesions also tend to be symmetrical and bilateral, distributed in the periventricular and deep white matter. MRI may also show lacunar infarcts and cerebral microbleeds.
To confirm the diagnosis of CADASIL, gene analysis was requested and the observed heterozygous missense variation in the patient was present in the EGF-like domain of the NOTCH3 protein. Though different missense (p.Cys49Tyr) affecting the same domain had previously been reported in patients affected with CADASIL, the (p.Cys49Trp) variant has not been reported in the Genome Aggregation Database—gnomAD (a collection of research articles and related content from the gnomAD Consortium that describe and analyze human genetic variation). The disease is associated with vascular smooth muscle degeneration primarily involving small vessels which is linked to mutations in the NOTCH3 gene (cysteine-altering pathogenic variants) whose product is a transmembrane receptor which are predominantly expressed in adults by vascular smooth muscle cells and pericytes. However, the pathogenesis of CADASIL still remains unclear [16].
Conclusion
As CADASIL has a relapsing and partially remitting course, patients may receive treatment for demyelination which may not be necessary. The presence of encephalopathy, migraine, and stroke in patients with relapsing–remitting illness should lead one to further probe into the family tree for a heritable neurological illness. In addition, knowledge of characteristic MRI findings seen in CADASIL can help discern the diagnosis.
Availability of data and materials
Not applicable.
Abbreviations
- ADC:
-
Apparent diffusion co-efficient
- ANA:
-
Antinuclear antibody
- CADASIL:
-
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy
- CSF:
-
Cerebrospinal fluid
- DWI:
-
Diffusion-weighted imaging
- FLAIR:
-
Fluid-attenuated inversion recovery
- MRI:
-
Magnetic resonance imaging
- MS:
-
Multiple sclerosis
- RRMS:
-
Relapsing–remitting multiple sclerosis
- TOF:
-
Time-of-flight
- WMH:
-
White matter hyperintensity
References
Wang MM. CADASIL. In: Geschwind DH, Paulson HL, Klein C, editors. Handbook of clinical neurology. Amsterdam: Elsevier; 2018. p. 733–43 (10.1016/B978-0-444-64076-5.00047-8).
Tournier-Lasserve E, Joutel A, Melki J, Weissenbach J, Lathrop GM, Chabriat H, et al. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy maps to chromosome 19q12. Nat Genet. 1993;3(3):256–9. https://doi.org/10.1038/ng0393-256.
Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, et al. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature. 1996;383(6602):707–10. https://doi.org/10.1038/383707a0.
Joutel A, Vahedi K, Corpechot C, Troesch A, Chabriat H, Vayssière C, et al. Strong clustering and stereotyped nature of Notch3 mutations in CADASIL patients. Lancet. 1997;350(9090):1511–5. https://doi.org/10.1016/S0140-6736(97)08083-5.
Peters N, Opherk C, Bergmann T, Castro M, Herzog J, Dichgans M. Spectrum of mutations in biopsy-proven CADASIL: implications for diagnostic strategies. Arch Neurol. 2005;62(7):1091–4. https://doi.org/10.1001/archneur.62.7.1091.
Dichgans M, Mayer M, Uttner I, Brüning R, Müller-Höcker J, Rungger G, et al. The phenotypic spectrum of CADASIL: clinical findings in 102 cases. Ann Neurol. 1998;44(5):731–9. https://doi.org/10.1002/ana.410440506.
Markus HS, Martin RJ, Simpson MA, Dong YB, Ali N, Crosby AH, et al. Diagnostic strategies in CADASIL. Neurology. 2002;59(8):1134–8. https://doi.org/10.1212/wnl.59.8.1134.
Viswanathan A, Gschwendtner A, Guichard JP, Buffon F, Cumurciuc R, O’Sullivan M, et al. Lacunar lesions are independently associated with disability and cognitive impairment in CADASIL. Neurology. 2007;69:172–9. https://doi.org/10.1212/01.wnl.0000265221.05610.70.
Ferrante EA, Cudrici CD, Boehm M. CADASIL: new advances in basic science and clinical perspectives. Curr Opin Hematol. 2019;26(3):193–8. https://doi.org/10.1097/MOH.0000000000000497.
Chabriat H, Joutel A, Dichgans M, Tournier-Lasserve E, Bousser MG. Cadasil. Lancet Neurol. 2009;8(7):643–53. https://doi.org/10.1016/S1474-4422(09)70127-9.
Uchino M. The pathomechanism and treatment of CADASIL. Rinsho Shinkeigaku. 2011;51(11):945–8. https://doi.org/10.5692/clinicalneurol.51.945.
Khan A, Abedi V, Li J, Malik MT, Esch M, Zand R. CADASIL vs. multiple sclerosis: is it misdiagnosis or concomitant? A case series. Front Neurol. 2020;11:860. https://doi.org/10.3389/fneur.2020.00860.
Aracki-Trenkic A, Stojanov D. Imaging characteristics of CADASIL patient with inherited and de novo gene NOTCH 3Q19 mutation. ECR 2014 Congress. 10.1594/ecr2014/C-0309
O’Sullivan M, Jarosz JM, Martin RJ, Deasy N, Powell JF, Markus HS. MRI hyperintensities of the temporal lobe and external capsule in patients with CADASIL. Neurology. 2001;56(5):628–34. https://doi.org/10.1212/wnl.56.5.628.
Auer DP, Pütz B, Gössl C, Elbel G, Gasser T, Dichgans M. Differential lesion patterns in CADASIL and sporadic subcortical arteriosclerotic encephalopathy: MR imaging study with statistical parametric group comparison. Radiology. 2001;218(2):443–51. https://doi.org/10.1148/radiology.218.2.r01fe24443.
Kalaria RN, Viitanen M, Kalimo H, Dichgans M, Tabira T, CADASIL Group of Vas-Cog. The pathogenesis of CADASIL: an update. J Neurol Sci. 2004;226(1–2):35–9. https://doi.org/10.1016/j.jns.2004.09.008.
Acknowledgements
Not applicable
Funding
The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
SR was the lead author in drafting and preparation of the case report. SA was the consultant in charge of the patient, was actively involved in decision-making as well as patient treatment, and contributed to the manuscript. PRPI and VCCK contributed, edited, and critically revised the final manuscript before submission. AJR contributed with the description and interpretation of the radio-diagnostic findings in the patient. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was in accordance with the ethical standards of the institutional ethical review committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent was taken from the patient for this study.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report along with its accompanying images.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rackimuthu, S., Ahmed, S., Ishwara, P.R.P. et al. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) masquerading as CNS demyelination. Egypt J Neurol Psychiatry Neurosurg 58, 67 (2022). https://doi.org/10.1186/s41983-022-00502-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41983-022-00502-9