
Abstract
Background
Thirty-six entomopathogenic fungi (EPF) were isolated from soil and insect cadaver samples, collected from different forest types, viz., wet evergreen, moist deciduous, dry deciduous and scrub type in South India. Partial sequences of two parsimony informative genes ITS and RPB1 were determined under a phylogenetic approach for assessing the genetic diversity.
Results
Twenty-seven RPB1 gene sequences and 34 sequences of ITS1, 5.8S and ITS2 regions belonging to 36 EPF were analysed for identification and characterization. Four species of Metarhizium viz., M. anisopliae, M. roberstii, M. majus and M. guizhouense were differentiated. The isolates could be grouped into four main clades of 1–5. Most of the fungi appeared to be closely related to M. anisopliae. Based on the colony characters, colour, conidial size and shape, 27 isolates were morphologically identified as M. anisopliae. Seven strains were apparently related to M. robertsii, three isolates were similar to M. majus and the remaining one was identified as M. guizhouense. Morphological studies in congruence with phylogenetic analysis resolved the species diversity. Bioassay studies showed that M. quizhouense, M. majus and M. robertsii were effective against the banana stem weevil Odoiporus longicollis.
Conclusions
This is the first attempt to study the diversity and occurrence of Metarhizium species in forests of South India. Wet evergreen forest of Aralam in South India was rich in EPF diversity particularly for three species namely, M. quizhouense, M. robertsii and M. anisopliae.
Similar content being viewed by others

Background
The genus Metarhizium in the family Clavicipitaceae includes a group of highly virulent entomopathogenic fungi (EPF) classified under Ascomycota, Hypocreales. Several species of Metarhizium are reasonably efficient in causing serious epizootics, thereby capable of regulating insect populations in nature to a good extent (Lacey et al. 1999). Species of fungi under Metarhizium have a ubiquitous distribution and are present in soils of different landscapes including the forest ecosystem (Lomer et al. 2001). Reports about the ability of many varieties of M. anisopliae to produce specific structures such as microsclerotia to escape from desiccation provide useful cues about the distribution of the fungus in soil. Since it is very infective, its distribution and survival in soils may have relevance to the population of their arthropod host species. Analysing the diversity of this group would therefore seem worthwhile for identifying potential native isolates for control of regional pests (Meyling and Eilenberg 2007).
Metarhizium species like M. anisopliae, M. flavoviridae, M. acridum and M. robertsii are well-documented due to their wide occurrence (Bischoff et al. 2009). Several species of EPF have been shown to play multiple roles in nature, ranging from antagonists of plant pathogens to rhizosphere associates, endophytes and possibly even as plant-growth-promoters (Barelli et al. 2018). Metarhizium species are isolated from infected insect cadavers or through Galleria soil bait methods (Keyser et al. 2015).
Lately, researchers have found through molecular phylogeny studies that there are several major evolutionary lines and that the taxonomic relationships at the base of the tree are poorly resolved (Driver et al. 2000). However, researchers (Kepler et al. 2014) could distinguish 20 different species of Metarhizium based on molecular phylogeny. Hence, within species, genetic diversity in particular habitats in the context of the revised taxonomy is largely unknown (Nishi et al. 2010). Even within a limited geographical area, considerable genetic variability of Metarhizium spp. can be found (Wyrebek et al. 2011).
Several M. anisopliae isolates were isolated from naturally infected insects and their diversity was studied through phylogenetic and comparative biological characters (Balachander et al. 2013) but it was based only on ITS sequences. Studies show that occurrence of cryptic groups of M. anisopliae subspecies lineage in South India were genetically distinctive, non-recombining and strongly associated with soil environments of two forest habitat types (Ramanujam et al. 2015).
The identification of these EPF using morphology-based identification keys will help in differentiating up to genus level, however differentiating the species within the same genus is very difficult. The use of multi-phylogenetic tree analysis of RNA polymerase II (RPB1) gene sequence will aid in identifying M. anisopliae, subspecies M. roberstii, M. majus and M. guizhouense (Kepler and Rehner 2013). RBP1 is the enzyme responsible for transcription of protein-coding gene of the largest subunit of this protein.
The pseudostem borer, Odoiporus longicollis Oliver (Coleoptera/Curculionidae) also known as banana stem weevil (BSW) can cause substantial damage in terms of production and productivity of bananas and plantains all over the world (Prasuna et al. 2008). Recent reports suggest that O. longicollis is distributed over diverse geographical locations of India (Thippaiah et al. 2011) and serious crop loss due to BSW has been reported from banana-growing states (Visalakshi et al. 1989). Since banana is a food crop, safer control methods of BSW are needed. One important safe alternative is use of EPF as they can grow naturally in soils and are capable of infecting various insects (Awasthi et al. 2017).. Researchers have tried to use M. anisopliae, Beauveria bassiana and their related species to control O. longicollis (Sivakumar et al. 2019) but with limited success.
Phylogenetic analyses is important to assess the relationship between molecular and physiological characters of the commonly occurring Metarhizium spp., including for the four varieties of M. anisopliae, which are taxonomically redefined as subspecies. In the present study, the genetic diversity of Metarhizium spp. occurring naturally in forest soils of South India was determined for the first time, using sequence analysis of the non-coding and protein-encoding regions of RPB1 and ITS region. Morphological and multi-phylogenetic analysis were carried out to decipher intra-species relationship. The isolated Metarhizium spp. were also assessed for their biological control potential in combating O. longicollis.
Methods
Collection of samples
Methodological surveys were made to various forests of South India encompassing the states of Tamil Nadu, Kerala, Karnataka and Andhra Pradesh (Table 2). Dead insects and soil samples were collected from these forests viz., wet evergreen, moist deciduous, dry deciduous and scrub forest (Table 2) and relevant GPS data obtained. Isolation of EPF was carried out at the laboratory of Division of Insect Ecology and Conservation, Ashoka Trust for Research in Ecology and the Environment (ATREE), Bengaluru (Karnataka) India. The sampling method was as per protocols previously described (Sánchez-Peña et al. 2011). A total of 300 soil samples were collected from randomly chosen sampling sites spread over 0.5–1 km2 area with an approximate distance of 20–25 m apart. Five samples were collected from each plot and pooled to make one sample, before collecting, the surface was cleared of litter and dried leaves. Samples (about 200 g) were collected up to a depth of 20 cm using a shovel and transferred to air tight polythene covers, labelled and sealed immediately to prevent loss of moisture. The soil samples were then transferred to 500-mL plastic containers and maintained in dark for 30 days.
Isolation and purification of Metarhizium spp. by insect baiting
Insect baiting was done by use of larvae of Galleria mellonella L. (sourced from ICAR-National Bureau of Agricultural Insect Resource, Bengaluru-24). Soil of 100 g, cleaned of roots and gravel, was placed in Petri dish (90 mm), slightly moistened and mixed well before transferring G. mellonella larvae. Galleria larvae were first surface-sterilized by dipping sequentially in 70% ethyl alcohol, 1% sodium hypochlorite, and sterile distilled water, each for 2–3 min. About ten third and fourth larval instars of G. mellonella were introduced into each dish. The Petri dishes were incubated under dark at room temperature (24–28 °C).
Based on active period for forest pests, extensive surveys were conducted during November 2013 to December 2015 in the forests of South India and naturally infected insect cadavers of teak defoliators Hyblaea puera (Cramer) and Hypsipyla robusta (Moore), Eutectona machaeralis (Walker), Paligama choeralis Walker; root grubs Otiorhynchus sulcatus (Fabricius), Protaetia aurichalcea (Fabricius) and Melolontha melolontha (Linnaeus) were collected (identified at ICAR-National Bureau of Agricultural Insect Resource-Bengaluru-24). Cadavers were dissected and placed on Potato Dextrose Agar (PDA) medium and incubated at 28 °C with 90% RH. Metarhizium spp. was further purified using Veen’s medium containing Dodine (Veen and Ferron 1966). Same isolation procedure was adopted for infected Galleria larvae.
Morphological characteristics and conidial measurements
After establishing the pure cultures, slide culture technique (Riddell 1950) was adopted and on the basis of microscopic analysis (Model Leica) of morpho-taxonomic characters like colony colour, pigmentation, spore size/shape, nature of phialides, mycelial mat, etc. (Kepler et al. 2014) and preliminary taxonomic assignments were given (Table 2).
Genomic DNA extraction, PCR amplification
DNA was extracted by CTAB method from 50 to 100 g of lyophilized mycelium of the fungus that was cultured in Potato Dextrose Broth for 5–7 days (Rogers and Bendich 1994). Extracted DNA was suspended in EB buffer (10 mM Tris–HCl, pH 8.5) and stored at − 20 °C until used. Total DNA concentration was measured in spectrophotometer at 280 nm. PCR amplification of the protein-coding region of RPB genes and non-coding regions of internal transcribed spacer (ITS) of the nuclear ribosomal DNA (18S, 5.8S and 28S rDNA) partial sequences was done as per earlier protocols (Curran et al. 1994; Stiller and Hall 1997; Entz et al. 2008; Bischoff et al. 2009) and detailed in (Table 1). PCR amplification was performed using a thermal cycler (Bio-Rad, T100TM). Each PCR reaction consisted of 25-μl reaction mixtures containing 1X Taq buffer, 0.4–0.6 mM of each primer, 0.2 mM of dNTP mix, 1U of Taq DNA polymerase (GeNei) and 20–50 ng/µl template DNA. Primers for RPB1 and ITS were as per previous protocols (Kepler and Rehner 2013). The PCR amplification had one initial cycle at 95 °C for 3 min, followed by 35 cycles of denaturation at 95 °C for 1 min, extension at 72 °C for 2 min and a final extension at 72 °C for 10 min. The PCR products were separated on 1% agarose gel containing 0.05% of EtBr in 0.5 × TBE buffer and visualized in a gel documentation system (model DNR, Israel). The PCR products were sequenced in an ABI 3130XL genetic analyser at (Chromous Biotech Pvt. Ltd., Bangalore).
Sequence analysis and construction of phylogenetic tree
The sequence chromatograms were manually corrected and aligned using Bio-Edit tools at National Centre for Biotechnology Information (NCBI) and the contigs were assembled using the program CAP3 (Huang and Madan 1999). The amplification of ITS regions was achieved for all 34 of our isolates and these sequences were submitted to NCBI. Before submitting, the sequences were edited as described above. Similarly for RPB1 gene, only 27 sequences were amplified from the 36 isolates. Hence, a total of 34 ITS + 27 RPB1 sequences were submitted to NCBI. Phylogenetic analysis was done separately for ITS and RPB1 sequences. For ITS, we used nine reference sequences in the phylogenetic analysis. Again for RPB1 gene, 11 reference sequences apart from the 27 of our own RPB1 sequences submitted to NCBI were used.
The evolutionary history was inferred by using the maximum likelihood method and Tamura 3-parameter model. To analyse both sequence data sets, the first 50% of the resulting trees were discarded (burn in), and a 70% majority rule consensus tree was then calculated from the remaining trees. Neighbour-joining (NJ) analysis with a maximum composite likelihood (MCL) option was also performed on the same alignment using MEGA X Huang and Madan (1999) and Kumar et al. (2018) branch support was estimated by bootstrap analysis (Felsenstein 1985) based on 10,000 bootstrap replicates.
Bioassay against Odoiporus longicollis
Laboratory rearing
Banana stem weevils were collected from banana growing areas of Tamil Nadu and Karnataka during 2015–2016. The weevils were maintained at Division of Genomic Resources, at ICAR-National Bureau of Agricultural Insects Resource, Bengaluru (Karnataka), India. Rearing of larvae was carried out in the laboratory on artificial diet for two generations (Priyadarshini et al. 2014). The pseudostems were changed once in 15 days and were placed in another plastic box containing moist filter paper disc. After hatching, the first instar larvae were transferred on to fresh pieces of pseudostem. Larvae were reared in various stages viz. Egg, first, second and third instar. Adult larvae were allowed to feed on leaf sheath at 28 ± 1 °C, 75 ± 5% RH. Subsequently, larvae were transferred to a moist sterile filter paper within an unsealed Petri dish (12 cm in diameter). Approximately 35 days later, laboratory-reared larvae were obtained for further analysis.
Preparation of spore suspension and bioassay
The bioassay against O. longicollis was carried out as per protocol of Padmanaban and Sathiamoorthy (2001). All the Metarhizium isolates were grown in Petri plates on potato dextrose agar (PDA) for 10 days at 28 °C with 70–80% RH. The mycelium was harvested by scraping the mycelial mat from PDA. The conidial suspension was prepared in sterile water containing 0.1% Tween-80, and the conidial concentration was determined using a haemocytometer. The suspension was then adjusted to 1 × 107conidia/ml. Five different conidial suspensions (1 × 103, 1 × 104, 1 × 105, 1 × 105 and 1 × 107conidia/ml) of all the isolates were prepared for bioassay. The insect bioassays were carried out using third instar larvae of banana stem weevil O. longicollis. For each test organism, a set of 30 larvae in triplicate were dipped individually in 10 mL conidial suspension for 5 s and each treated larva was individually transferred to a separate sterile vial containing moist Whatman filter paper No. 1 and a piece of disinfected castor or teak leaf that was changed on alternate days. The experiment was laid in a randomized complete block design (RCBD) with three replications. Mortality was recorded consecutively for eight days.
Statistical analysis
The data of mortality was subjected to probit analysis by using the Proc Probit procedure of the SPSS statistical package (SPSS Institute 2008), and the median lethal concentration (LC50) needed to kill at least 50 per cent of the population was determined.
Results
A total of 36 EPF were isolated from the 300 soil and cadaver samples collected from forest regions of South India during 2013–2016 (Table 2). Soil moisture contents ranged from 6.4 to 47.42%, and the EPF were initially identified based on morphological keys including conidial shape/size, and colony morphology. Nineteen Metarhizium spp. were isolated through Galleria bait but unresolved as separate monophyly and genealogy. We designated them as M. anisopliae until further characterization.
Among the 36 isolates, 27 were morphologically similar to M. anisopliae based on their colony, conidial colour, size and shape. Other seven strains were apparently related to M. robertsii, further three isolates were similar to M. majus and the remaining one appeared to be M. guizhouense. M. anisopliae produced green conidia (some near brown) without radial sulcations, whereas M. robertsii produced conidiophores that were branched, erect, closely or loosely grouped, forming sporulation layer. Isolates belonging to M. guizhouense had phialides borne singly or in pairs and sometimes in whorls. Isolates showing dark green conidia (some near brown), and generally not expressing heavy radial sulcations, were classified as M. majus.
Phylogenetic tree analyses of ITS region
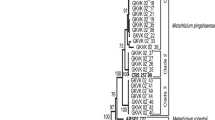
Phylogenetic tree was constructed using Neighbour-Joining tree (MEGA-X) bioinformatics software tool, and the genetic relationships between the isolates were analysed. The dendrogram shows the genetic relatedness among the algorithm in the Metarhizium spp. (Fig. 1). This analysis involved 45 nucleotide sequences (34 of our own plus 11 reference sequences). All positions containing gaps and missing data were eliminated (complete deletion option). There were a total of 371 positions in the final dataset.

Tree topology based on ITS sequence. The phylogeny of Metarhizium spp. within the complex of M. anisopliae, M. robertsii, M. majus and M. quizhouense (clades 1–5) constructed from the 5′ end of the ITS region analysis involving 45 nucleotide sequences classified into the clade in South India. The scale bar indicates evolutionary distance
The partial ITS1 and ITS2 region sequences of 27 M. anisopliae isolates showed 95–100 per cent homology at 470–570 bp (Table 1) and majority of them were grouped into major clade-1, thus establishing the monophyly. Six other isolates of M. anisopliae were differentiated in clade 2 indicating minor variation from the parent population. The 20 M. anisopliae isolates clustered as clade 1 showed 98% commonality with the reference M. anisopliae (CBS127632), and out of these 20 isolates, 14 were from Galleria bait. The other six that were isolated from infected insect cadavers also showed distinct similarity. The spore morphology of M. anisopliae isolated from soils of Karnataka (Bannerghatta, Nallal and Jarakbande) was bigger in size but could be morphologically distinguished.
Seven isolates of M. robertsii clustered in clade 4 with significant bootstrap. Interestingly, all these M. robertsii isolates were isolated from naturally infected cadavers of Protaetia aurichalcea and Otiorhynchus sulcatus that were collected from single location (Aralam) indicating a common ancestral origin and split. However, isolates of M. anisopliae were found to be ubiquitous in distribution. Thus, the species complexes share a well-supported sister relationship of significant diversity. There were seven isolates of M. robertsii identified in this study that had 100 per cent analogy within 537–687 bp range (Table 1) and represented 20.2% of area covered.
The phylogenetic tree for ITS showed one M. majus isolate branched in clade 3, and this isolate was sourced from infected cadavers of Melolontha melolontha (Table 2). The other isolate that clustered in clade 3 was identified as M. guizhouense and this EPF was isolated from infected cadavers of Eutectona machaeralis. Nearly, 100% homology was seen in M. majus and M. quizhouense in 572 bp and 558 bp range (Table 1). Hence, these two Metarhizium species could be closely related. Thus, the phylogenetic analyses clustered M. majus and M. quizhouense as separate and they showed 89% homology with NCBI reference sequences of M. majus (ARSEF1946) and M. guizhouense (ARSEF977). M. brunneum (ARSEF820) was represented as out group for sister species (Fig. 1).
The genealogy of the unique isolate M. quizhouense that showed significant diversity was identified within M. anisopliae. No EPF were found in laterite and sandy soils that represented scrub forest types of Ramanagara, Mysore, Madurai and Hindupur areas. These soils tend to have lower water content and pH and hence do not support survival of EPF.
Six reference sequences clustered in clade 5. The three identified isolates in clade 4 (Fig. 1) showed slight similarity to the reference M. brittlebankisoides (UAMH9198) clustered in clade 5. Other sister reference species in clade 5 were M. frigidum (ARSEF4124) and M. flavoviride (BC1163). As a final point, phylogenetic tree was rooted using out groups Lecanicillum lecanii (NBAII-Vl-15), Cordyceps bassiana (BCC2119) and Beauveria bassiana (BbH51Z02) and were used as a sister to the phylogenetic analysis (Fig. 1).
Phylogenetic analysis of the RPB1 gene sequences of Metarhizium spp.
Phylogenetic analysis of the RPB1 gene sequences yielded compatible trees that each resolved five well-supported terminal lineages or branches (Fig. 2). The results are summarized showing the maximum likelihood method with support values for species clades and the analysis involved 38 nucleotide sequences (including 11 reference sequences). The data aided in identification of Metarhizium species and are grouped into clades 1–6 according to their sequence and congruence to the classification proposed earlier (Table 2, Fig. 2).

Tree topology based on RPB 1 sequence. The phylogeny of Metarhizium spp. within the complex of M. anisopliae, M. robertsii and M. majus (clades 1–6) constructed from the 5′ end of the RPB1 gene analysis involving 38 nucleotide sequences were classified into the clade in South India. Strict (100%) consensus tree of the RPB1 phylogenies determined in the maximum composite likelihood (MCL) analysis. The scale bar indicates evolutionary distance
Analysis of RPB1 sequences indicated sequence similarity with three recognized species of M. anisopliae, M. roberstii and M. majus. The partial 530 bp (Table 1), RPB1A gene sequences showed 95–100% homology in 17 M. anisopliae isolates. Sequence analysis of 525 bp amplified (Table 1) RPB1B primers showed 100% homology with seven isolates and were identified as M. robertsii. The amplified 490 bp (Table 1) RPB1C gene sequence had 99% homology with M. majus and is described for three isolates. All 27 RPB1 nucleotide sequences amplified in our EPF isolates were submitted to NCBI (Table 2).
The phylogenetic tree produced from RPB1 gene sequences divided the majority of M. anisopliae strains into two major clades (4 and 5) with marginal support of 100% (Fig. 2) from the 17 native isolates of M. anisopliae (Table 2). The most interesting feature was that nine native strains of M. anisopliae were associated with Lepidoptera hosts H. robusta and G. mellonella (Table 2). The other eight M. anisopliae isolates were grouped together in clades 4 and 5 with significant bootstrap (99 and 90%), followed by closely related seven isolates of M. robertsii that clustered in clade 2 with the reference sequences of M. robertsii (AGF65 and AGF658). Therefore, M. robertsii isolates that were morphologically distinct with olive green conidia and white to yellow mycelia represents highly complex cryptic phylogenetic species.
The three indigenous M. majus isolates and the reference strain M. majus (AREF1914) were placed basically in clade 3 and continued with significant bootstrap support (> 90% homology). The closely related reference sister species viz., M. acridum (AGF159), M. frigidum (BCF34), and M. lepidiotae (AGF252) were used as a sister to the Metarhizium monophyly in clade 1 (Fig. 2). The phylogenetic tree was rooted using reference out groups (clade 6) Beauveria bassiana (BUB257), Cordyceps brongniartii (NBRC101395) and Lecanicillium lecanii (C42). The relatively large number of informative characters may explain the marginal BioNJ algorithms to a matrix of pairwise distances estimated using the MCL approach.
Determination of median lethal concentration (LC50) against Odoiporus longicollis
The virulence of 28 strains of Metarhzium spp. was investigated against the banana stem weevil O. longicollis and it was observed that all the M. quizhouense (ArMz1W) isolates were equally effective and the LC50 values ranged between 1 × 103 to 1 × 107 conidia/ml (Table 3).
Probit analysis based on conidia concentration and mortality response at 8-day post-inoculation (Table 3) indicated that M. quizhouense (ArMz1W) showed low LC50 value (8.436 × 105 conidia/ml), followed by M. anisopliae (10.411 × 105 conidia/ml) indicating that M. quizhouense was more virulent. Surprisingly, M. robertsii (ArMz3R) was also virulent with LC50 value of 11.525 × 103 conidia/ml.
Discussion
Currently, high degree of genetic diversity is reported among the individuals of the naturally occurring Metarhizium fungi from different regions and habitats across the tropical regions of South India (Humber 1997). Researchers have traditionally utilized morphological characteristics to distinguish among species of Metarhizium (Rangel et al. 2010; Fernandes et al. 2011). Phenotypic characteristics (viz., conidia colour, colony shape and size) seem to be correlated with genotype; and they may help in categorizing new Metarhizium isolates. The observed morphological characters of obtained Metarhizium spp. were in accordance with the reference species described earlier (Velavan et al. 2017). Unfortunately, morphology is still used as key for species identification of the M. anisopliae complex even though an individual isolate may show varied morphological features under varying environmental and physiological conditions (Fernandes et al. 2011). In India, identification of Metarhizium spp. is also based mostly on morphological features (Ravindran et al. 2015).
Hence, molecular data with morphology would be a fairly rational approach in resolving taxonomic complexities in the Metarhizium lineage (Kepler et al. 2014). Therefore, sequence analysis of the non-coding and protein-encoding regions of RPB1 and also ITS region were carried out with construction of phylogenetic tree and analyses.
The biodiversity of EPF in tropical ecosystems is not fully investigated (Rocha et al. 2013) The objective of this study was to isolate and identify Metarhizium spp. present in undisturbed soils of South India and test their biocontrol potential. The results established a molecular framework for taxonomic, phylogenetic and comparative biological investigations of entomopathogenic fungi isolated from naturally infected insects. Based on the partial sequences submitted to GenBank 36 isolates were identified with 97–100% similarity and 27 of them were M. anisopliae, seven belonged to M. robertsii, three as M. majus and one identified as M. guizhouense. Studies show that forest ecosystems had less occurrence and diversity of Metarhizium spp. but were more abundant in agriculture soils (Schneider et al. 2012). The density and diversity of soil organisms in natural habitats such as forests differs profoundly than in arable land (Meyling et al. 2009). Highest incidences of Metarhizium spp. were observed when G. mellonella was used as bait (Keyser et al. 2015). They suggested that isolation from infected insect cadavers will determine the degree of success in isolating host-specific EPF.
It is essential to analyse the genetic diversity of Metarhizium in ecological studies in order to resolve the status of several species and varieties in the Metarhizium lineage, especially of several cryptic species. Such studies are important for identifying location/region specific species/isolates for exploitation in biological control (Meyling and Eilenberg 2007). As in several of the studies cited above, all the gene partitions that were analysed support the delimitation of each Metarhizium species recognized herein. However, a minimum threshold of two-gene support was achieved for the recognition of seven phylogenetic species (Lopes et al. 2013). Where a two-gene minimum support threshold was acquired in M. majus and M. guizhouense with fixed differences in conidial morphology and a single-gene phylogeny was used as the basis for their recognition. Congruence of only one sequenced gene and either phenotype or geographic endemism is commonly invoked as the basis to propose species status in fungi (Kepler et al. 2014).
This is the first attempt to study the diversity and occurrence of M. anisopliae, M. roberstii, M. majus and M. guizhouense in forests of South India. Phylogenetic diversity and inter relationship of sequence analysis of the ITS and RPB1 sequence data of 36 isolates were grouped into the lineage of M. anisopliae, subspecies: M. roberstii, M. majus and M. guizhouense. Spores growing on cadavers survived at 4 °C as long as they were kept dried and also being frozen for future use in bioassay and gene expression studies.
Obtained results also suggest that the conidial sizes of M. anisopliae, M. roberstii, M. guizhouense, and M. majus were in congruent with the phylogeny based on sequences of the 5′ end of ITS region and RPB1 gene. Further research may prove the congruence between coding region EF1α and EF1α (intron and exon) phylogeny as well conidial sizes, but it is possible that the morphology is indifferent to phylogeny because the conidial size of fungi changes because of polyploidy. It is necessary to directly examine the polyploidy of M. anisopliae, M. roberstii, M. guizhouense and M. majus to gain further understanding of the relationship between the morphology and phylogeny.
Formulations of B. bassiana and M. anisopliae exhibited LC50 value of 1.25 × 106 conidia/mL against O. longicollis at 15 days (Sivakumar et al. 2019). In the present studies, M. quizhouense (ArMz1W) exhibited LC50 figure of 8.436 × 105 conidia/ml in 8 days indicating that it could be an effective biocontrol agent against O. longicollis.
Conclusions
In the present study, an attempt was made to explore the occurrence and diversity of Metarhizium fungi in the soils across different forest habitats. Knowledge about their ecological host range and effects of biotic and abiotic factors on their occurrence and distribution will help exploit them in the strategy of conservation biological control. Aralam wet evergreen forest of South India was rich in EPF diversity, particularly for the three species, namely M. quizhouense, M. robertsii and M. anisopliae. Data on diversity of EPF occurring in forest soils of south India is not available. Bioassay against O. longicollis showed that M. guizhouense and M. robertsii were effective suggesting that these could be deployed in field. This is the first report showing the toxic nature of M. anisopliae subspecies against O. longicollis.
Availability of data and materials
All data of the study have been presented in the manuscript, and high quality and grade materials were used in this study.
Code availability
(MEGA X, CAP3, NCBI and SPSS) software was used in the paper for statistical analysis.
Abbreviations
- PDAY:
-
Potato dextrose agar yeast extract
- TBE:
-
Tris/Borate/EDTA)
- EDTA:
-
Ethylenediaminetetraacetic acid
- NaOCl:
-
Sodium hypochlorite
- CTAB:
-
Cetyltrimethylammonium bromide
- NCBI:
-
National Center for Biotechnology Information
- EPF:
-
Entomopathogenic fungi
- ITS:
-
Internal transcribed spacer
- RPB1:
-
RNA polymerase II largest subunit
- BSW:
-
Banana stem weevil
- IPM:
-
Integrated pest management
- LC:
-
Lethal concentration 50%
- RCBD:
-
Randomized complete block design
References
Awasthi NS, Sridharan S, Mohankumar S (2017) In Vitro evaluation of native isolate of Metarhizium anisopliae (Metchinkoff) Sorokin and its oil in water formulations against Odoiporus longicollis Olivier. J Biol Control 31(4):248–252
Balachander M, Sasidharan ROK, TO, (2013) Dissemination of Metarhizium anisopliae infection among the population of Odentotermes obesus (Isoptera: Termitidae) by augmenting the fungal conidia with attractants. J Asia-Pac Entomol 16:199–208
Barelli L, Moreira CC, Bidochka MJ (2018) Initial stages of endophytic colonization by Metarhizium involves rhizoplane colonization. Microbiology 164:1531–1540
Bidochka MJ, Kamp AM, Lavender TM, de Koning J, de Croos JNA (2001) Habitat association of two genetic groups of the insect-pathogenic fungus Metarhizium anisopliae: uncovering cryptic species. Appl Environ Microbiol 67:1335–1342
Bischoff JF, Rehner SA, Humber RA (2006) Metarhizium frigidum sp. nov: a cryptic species of M. anisopliae and a member of the M. flavoviride complex. Mycologia 98:737–745
Bischoff JF, Rehner SA, Humber RA (2009) A multilocus phylogeny of the Metarhizium anisopliae lineage. Mycologia 101:512–530
Curran J, Driver F, Ballard JWO, Milner RJ (1994) Phylogeny of Metarhizium: analysis of ribosomal DNA sequence data. Mycol Res 98:547–552
Driver F, Milner RJ, Trueman JWH (2000) A taxonomic revision of Metarhizium based on a phylogenetic analysis of rDNA sequence data. Mycol Res 104:134–150
Entz SC, Kawchuk LM, Johnson DL (2008) Discovery of a North American genetic variant of the entomopathogenic fungus Metarhizium anisopliae var. anisopliae pathogenic to grasshoppers. Biol Control 53:327–339
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the boostrap. Evolution 39(4):783–791
Fernandes EKK, Keyser CA, Rangel DEN, Foster RN, Roberts DW (2011) CTC medium: a novel dodine-free selective medium for isolating entomopathogenic fungi, especially Metarhizium acridum, from soil. Biol Control 54:197–205
Huang XA, Madan A (1999) CAP3: a DNA sequence assembly program. Genome Res 9(9):868–877
Humber RA (1997) Fungi: identification. In: Lacey LA (ed) Manual of techniques in insect pathology. Academic Press, San Diego, pp 153–185
Kepler RM, Rehner SA (2013) Genome-assisted development of nuclear intergenic sequence markers for entomopathogenic fungi of the Metarhizium anisopliae species complex. Mol Ecol Resour 13(2):210–217
Kepler RM, Humber RA, Rehner SA (2014) Clarification of generic and species boundaries for Metarhizium and related fungi through multigenephylogenetics. Mycologia 106(4):811–829
Keyser CA, Henrik H, Licht DF, Steinwender BM, Meyling NV (2015) Diversity within the entomopathogenic fungal species Metarhizium flavoviride associated with agricultural crops in Denmark. BMC Microbiol 15:249
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35(6):1547–1549
Lacey LA, Kirk AA, Millar L, Mercadier G, Vidal C (1999) Ovicidal and larvicidal activity of conidia and blastospores of Paecilomyces fumosoroseus (Deuteromycotina: Hyphomycetes) against Bemisia argentifolii (Homoptera: Aleyrodidae) with a description of a bioassay system allowing prolonged survival of control insects. Biocontrol Sci Technol 9:9–18
Lomer CJ, Bateman RP, Johnson DL, Langewald J, Thomas M (2001) Biological control of locusts and grasshoppers. Annu Rev Entomol 46:667–702
Lopes RB, Martins I, Souza DA, Faria M (2013) Influence of some parameters on the germination assessment of mycopesticides. J Invert Pathol 112(3):236–242
Meyling NV, Eilenberg J (2007) Ecology of the entomopathogenic fungi B. bassiana and M.anisopliae in temperate agro-ecosystems: potential for conservation biological control. Biol Control 43:145–155
Meyling NV, Lübeck M, Buckley EP, Eilenber GJ, Rehner SA (2009) Community composition, host range and genetic structure of the fungal entomopathogen Beauveria in adjoining agricultural and semi-natural habitats. Mol Ecol 18:1282–1293
Nishi O, Iiyama K, Yasunaga-Aoki C, Shimizu S (2010) Incongruence between EF-1α Phylogeny and Morphology of Metarhizium majus and Metarhizium guizhouense in Japan. Entomotech 34:19–23
Padmanaban B, Sathiamoorthy S (2001) The banana stem weevil Odoiporus longicollis. Musa Pest Fact Sheet No.5. INIBAP, Montpellier, France
Prasuna AL, Jyothi KN, Prasad AR, Padmanaban YJS, B, (2008) Olfactory responses of banana pseudostem weevil, Odoiporus longicollis Olivier (Coleoptera: Curculionidae) to semiochemicals from conspecifics and host plant. Curr Sci 94(7):896–900
Priyadarshini GI, Mukherjee U, Kumar N (2014) Biology and seasonal incidence of pseudostem weevil, Odoiporus longicollis Oliver (Coleoptera: Curculionidae) in banana. Pest Manag Hortic Ecosys 20(1):8–13
Ramanujam B, Poornesha B, Yatish KR, Renuka S (2015) Evaluation of pathogenicity of different isolates of Metarhizium anisopliae (Metchnikoff) sorokin against maize stem borer, Chilopar tellus (Swinhoe) using laboratory bioassays. Biopestic Int 11(2):0973-483X/11/89-95.
Rangel DEN, Dettenmaier SJ, Fernandes EKK, Roberts DW (2010) Susceptibility of Metarhizium spp. and other entomopathogenic fungi to dodine-based selective media. Biocontrol Sci Technol 20:375–389
Ravindran K, Qiu D, Sivaramakrishnan S (2015) Sporulation characteristics and virulence of Metarhizium anisopliae against subterranean termites (Coptotermes formosanus). Int J Microbiol Res 6(1):01–04
Riddell RW (1950) Permanent stained mycological preparations obtained by slide culture. Mycologia 42:265–270
Rocha LN, Inglis PW, Humber RA, Kipnis A, Luz C (2013) Occurrence of Metarhizium spp. in Central Brazilian soils. J Basic Microbiol 53:251–259
Rogers SO, Bendich AJ (1994) Extraction of total cellular DNA from plants, algae and fungi. Plant Mol Biol Man 1(8):183–190
Sánchez-Peña SR, Lara SJ, Medina RF (2011) Occurrence of entomopathogenic fungi from agricultural and natural ecosystems in Saltillo, Mexico, and their virulence towards thrips and whiteflies. J Insect Sci 11:1
Schneider S, Widmer F, Jocot K, Kolliker R, Enkerli J (2012) Spatial distribution of Metarhizium clade 1 in agricultural landscapes with arable land and different semi-natural habitats. Appl Soil Ecol 52:20–28
Sivakumar T, Jiji T, Naseema A (2019) Effect of pesticides used in banana agro-system on entomopathogenic fungus, Metarhizium majus Bisch, Rehner and Humber. Int J Trop Insect Sci 40:283–291
SPSS Inc (2008) SPSS Statistics for Windows, Version 17.0. SPSS Inc, Chicago
Stiller JW, Hall BD (1997) The origin of red algae: Implications for plastid evolution. Proc Natl Acad Sci USA 94:4520–4525
Thippaiah M, Ashok Kumar CT, Shivaraju C, Sudhirkumar S, Naveena NL (2011) Study of biology of banana pseudostem weevil, Odoiporus longicollis olivier. Int J Entomol 02(1):01–05
Veen KH, Ferron P (1966) A selective medium for the isolation of Beauveria tenella and of Metarhizium anisopliae. J Invert Pathol 8:268–269
Velavan V, Sivakumar G, Rangeswaran R, Sasidharan TO, Sundararaj R (2017) Metarhizium majus and Metarhizium robertsii show enhanced activity against the coleopteran pests Holotricha serrata L. and Oryctes rhinoceros L. J Biol Control 31(3):135–145
Visalakshi A, Nair GM, Beevi SN, Amma AMK (1989) Occurrence of Odoiporus longicollis (Olivo) (Coleoptera: Curculionidae) as a pest of banana in Kerala. Entomon 4(3):367–368
Wyrebek M, Huber C, Sasan RK, Bidochka MJ (2011) Three sympatrically occurring species of Metarhizium show plant rhizosphere specificity. Microbiology 157:2904–2911
Acknowledgements
Thanks are also due to the Director Ashoka Trust for Research in Ecology and the Environment (ATREE), Institute of Wood Science and Technology, Forest and wood protection, Bengaluru, (Karnataka) and ICAR-National Bureau of Agricultural Insect Resources, Bengaluru, Karnataka, India for providing facilities to undertake the study. The permission granted by the PCCF Karnataka and PCCF Kerala to undertake survey in the states is also acknowledged.
Funding
The authors are grateful to the Department of Biotechnology, New Delhi (BT/PR/13913/AGR/05/507/2010) for providing financial support to carry out this work (2013–2016) and Isolation and collection of EPF strains (ATREE).
Author information
Authors and Affiliations
Contributions
VV and TOS and RS performed the technical characterization on strains and drafted the manuscript. RR, AK and GS conceived the study and aided to draft the manuscript. VV conceived the study, participated in its design and coordination, and helped to draft the Manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Consent for publication was obtained from all individual participants included in the study.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in the published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Velavan, V., Rangeshwaran, R., Sivakumar, G. et al. Occurrence of Metarhizium spp. isolated from forest samples in South India and their potential in biological control of banana stem weevil Odoiporus longicollis Oliver. Egypt J Biol Pest Control 31, 131 (2021). https://doi.org/10.1186/s41938-021-00476-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41938-021-00476-5