
Abstract
The strength of sequence-related amplified polymorphism (SRAP) in comparison to random amplified polymorphic DNA (RAPD) in determining the genetic polymorphism among seven species of entomopathogenic nematodes (Steinernematidae) was tested. Nine RAPD and 12 SRAP primer pair’s combinations were used. The number of polymorphic bands and the polymorphism percentages was high in SRAP analysis (97 out of 107 bands were polymorphic (90.6%)) compared to RAPD (65 out of 89 bands were polymorphic (73%)). The highest number of RAPD bands was recorded for OPE-A-07 primer (14 band) while OPE-D-20 was scored the lowest band number (7 bands). The SRAP primers Me3-em2 registered the highest number of bands (13 bands) while Me3-em3and Me2-em2 showed the lowest band number (6 bands). The genotype-specific SRAP and RAPD markers for the different ENP species were recorded. The highest number of SRAP specific markers (7 markers) was scored for Steinernema glaseri, then 4 markers for S. abbasi 2 followed by S. riobrave (3 markers), 1 marker for S. abbasi 1, S. riobrave recorded the highest number of specific RAPD marker (4 specific markers) followed by S. carpocapsae DD 136 and S. glaseri (two specific markers), then only 1 specific marker for S. abbasi 1. Based on the data obtained from RAPD and SRAP analysis, the dendrogram was created to clarify the genetic distances among different species of the studied entomopathogenic nematodes. The present study indicated that SRAP was more powerful than RAPD analysis for detecting the genetic polymorphism among closely related species of nematodes. The genotype-specific markers detected from both RAPD and SRAP can be used in future biological control programs.
Similar content being viewed by others

Background
Entomopathogenic nematodes (EPNs) are a promising candidate for biological control of insects due to their ability to find hosts, integrity to non-target organisms and the environment, high reproductive potential, capacity to be mass-produced and to be used with other agrochemical pesticides and fertilizers (Laznik and Trdan 2014). Two EPN families, Heterorhabditidae and Steinernematidae, have separately evolved collaborative associations with insect pathogenic bacteria (Photorhabdus and Xenorhabdus) (Adams et al. 2006). Morphological characterizations cannot be used unambiguously to rank new isolates into a particular species; the tendency of using these nematodes as biological control agents depends upon the appliance required for rapid and accurate methods to determine the genetic diversity among existent populations of ENPs species (Hashmi et al. 1996). Molecular markers have been used to study genetic diversity; it mainly exposes naturally occurring polymorphisms at DNA level that makes it a powerful tool to determine the effectiveness of various factors on genetic diversity and population structure (Li et al. 2014). DNA markers are numerous and separate and are widespread throughout the genome. They are highly polymorphic and stable and allow organism identification in each developmental stage (Agarwal et al. 2008). Random amplified polymorphic DNA (RAPD), amplified fragment length polymorphism (AFLP), simple sequence repeats (SSR), inter-simple sequence repeats (ISSR), single-strand conformation polymorphism (SSCP), and sequence-related amplified polymorphism (SRAP) are highly polymorphic and constant and have been widely employed in molecular taxonomy, gene cloning, phylogeny, and genetic mapping, (Gasser et al. 2006; Mutlu et al. 2008; Zhao et al. 2009). RAPD has been used for the genetic variability and phylogenetic studies of different organisms including insects, EPNs, animals, and plants (Moghaieb et al. 2004). RAPD analysis was used for the characterizations of a new species of Heterorhabditis from Hawaii (Gardner et al. 1994). SRAP is known as a useful molecular marker system based on its reproducibility and low cost (Li and Quiros 2001). Currently, SRAP has been successfully utilized to evaluate genetic diversity in species. It has been shown to be more instructive than other PCR-based techniques in discovering genetic diversity (Budak et al. 2004) and has been used to study the genetic diversity and relationships among several species (Abedian et al. 2012).
As there is no data about the application of SRAP analysis in EPN identification and taxonomy, this study aimed to compare the accuracy of SRAP and RAPD analyses in detecting the genetic variations among seven EPN species from Steinernematidae.
Materials and methods
Nematodes culture
Seven steinernematid species were used, and their names and geographical origins are listed in Table 1.
According to the method of Dutky et al. (1964), the EPNs were cultured, using the last instar larvae of the greater wax moth (Galleria mellonella L.) as a host.
Extraction of DNA
Total genomic DNA was extracted according to the method of Kary et al. (2009) as follows:
The nematode (5000 IJ) were crushed in 15 μl 1 × PCR buffer and transferred to a sterilized 0.2-ml tube containing 10 μl of the same buffer. The tube was incubated at − 70 °C for 15 min and thawed at 60 °C then inoculated with 2 μl of 60 μg ml−1 proteinase K. The tube was incubated at 65 °C for 2 h and then heated at 95 °C for 15 min. After centrifugation at 16.000 rpm for 15 min., the supernatant including nematode DNA was collected and stored at − 70 °C until use. The quality and quantity of DNA were determined using 0.8% agarose gel electrophoresis and spectrophotometer.
RAPD analysis
PCR analysis
PCR reactions mixture containing 200 μM dNTPs, 1 μM of 9 arbitrary 10-mer primers (Operon Technology, Inc., Alameda, CA, USA), 10 ng DNA, 0.5 units of Red Hot Taq polymerase (AB gene House, UK) and 10 × Taq polymerase buffer (AB gene House, UK), and total volume are adjusted to 20 μl. For DNA amplification, Biometra thermal cycler (2720) was programmed as follows: 94 °C for 5 min, followed by 35 cycles of 94 °C for 1 min, 35 °C for 1 min, 72 °C for 1 min, and 72 °C for 7 min. The products were analyzed by electrophoresis in 1% agarose in TAE buffer, stained by ethidium bromide, and photographed under UV light. The sequence of the tested primers is listed in Table 2.
SRAP analysis
PCR reactions were performed in a total volume of 20 μl containing 2 μl of the DNA suspension, 2 μl of 10 × PCR buffer, 2 μl of MgCl2 (25 mM), 1 μl of dNTP mixture (10 mM of each dNTP), 0.5 U of Taq DNA polymerase, 10.5 μl of dd H2O, and 1 μl of each primers. PCR program includes 5 min of initial denaturation at 94 °C. The first 5 cycles were run at 94 °C, 1 min; 35 °C, 1 min; and 72 °C, 1 min, for denaturing, annealing, and extension, respectively. Then, the annealing temperature was raised to 50 °C for another 35 cycles (Li and Quiros 2001), and the sequence of the primers is listed in Table 3.
Band scoring and cluster analysis
Gel Doc 2000 Bio-Rad system was used for RAPD and SRAP, and gel image scanning and One Software v 4.0.1 (Bio-Rad Laboratories, Hercules, Co., USA) were used for analyses with quantity. To calculate the pairwise differences matrix and plot the dendrogram among the seven EPN species, the Systat ver. 7 computer program was used. Cluster analysis based on similarity matrices obtained with the unweighted pair group method using the arithmetic average (UPGMA) to estimate the dendrogram.
Results and discussion
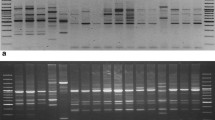
RAPD markers were used to study genetic diversity and population dynamics of various isolates in Heterorhabditidae and Steinernematidae (Hashmi and Gaugler 1997; Moghaieb et al. 2004). Nine random RAPD primers were used to evaluate the genetic polymorphism among the seven EPN species which generated 65 bands, 89 (73%) of them were polymorphic. The fragment size ranged between 110 and 1500 bp approximately, and the OPE-A-07 primer scored the highest number of RAPD bands (14 bands) while OPE-D-20 scored the lowest band number (7 bands) (Fig. 1, Table 4). Table 5 shows the specific RAPD marker for different species, and two specific markers were recorded for S. abbasi 1, S. riobrave, and S. glaseri followed by S. carpocapsae DD 136 (one specific marker), while none for S. abbasi 2, S. carpocapsae all, and S. scapterisci.

Genetic polymorphism among seven species belongs to the family Steinernematidae as revealed by RAPD analysis. M, 1 kbp plus DNA ladder; 1–7, the nematode species (1, S. abbasi 1; 2, S. abbasi 2; 3, S. carpocapsae DD 136; 4, S. carpocapsae all; 5, S. riobrave; 6, S. scapterisci; 7, S. glaseri)
RAPD has been used to explore the genetic diversity among species and strains of EPNs (Hashmi and Gaugler 1998), to define phylogenetic relationships (Liu and Berry 1996), and complement descriptions of new species (Stock et al. 1996). Padmanaban et al. (2014) developed RAPD markers to discriminate the genetic diversity in 25 recovered EPNs (Steinernema spp. and Heterorhabditis spp.) and found that polymorphism percentage that recorded in all the strains was 12.31%. The obtained data agrees with Ibrahim (2009), and by using RAPD analysis, the genetic polymorphism reached 96.9% and 91.4% between the nematode species belonging to Steinernema and Heterorhabditis, respectively. The genetic diversity among the EPN species from the genus Steinernema showed a high resemblance of shared DNA fragments between S. riobrave and S. scapterisci (95.4%) followed by S. scapterisci and S. carpocapsae all (89.4%). Hashmi et al. (1996) obtained similar data, where they screened 80 arbitrary 10-mer random primers for determining genetic variability among isolates of Heterorhabditis. Cluster analysis for Steinernema species showed that the cluster was divided into two branches, the first one was S. feltiae and the second was subdivided into two branches, one had S. carpocapsae and the other had S. glaseri. Shapiro et al. (1997) used RAPD-PCR to compare genetic variation in two strains of Heterorhabditis bacteriophora and had found a similar genetic heterogeneity of isolated population of H. bacteriophora to that reared in the laboratory for a long period. Moghaieb et al. (2004) employed protein banding pattern (SDS-PAGE) and RAPD markers to discriminate between two heat tolerant EPN isolates (HTS1 and HTS 2) and their original strain ISK-2, and nine arbitrary 10-mer primers were used. A total of 5 (21.7%) and 18 (39.1%) polymorphic bands were found out for protein and RAPD, respectively. They found that the EPN-specific markers represent 14% of the total markers detected by both analyses and 70% of them were RAPD markers, and these markers are recommended in providing a quick and reliable discrimination technique, compared to the protein analyses, to identify and distinguish between the heat tolerance and other EPN isolates.
SRAP is known as a useful molecular marker system, based on its propagation, low cost, and little knowledge that is necessary for target sequences in a species (Li and Quiros, 2001). The distinguishing power of molecular markers has been arranged as SRAP > SSR > ISSR > RAPD (Budak et al. 2004).
In this study, 12 different SRAP primer pairs among seven species of tested EPN were used. The data revealed that 97 bands out of 107 were polymorphic (90.6%) (Fig. 2, Table 6). The Me3-em2 primer recorded the highest number of bands (13 bands) followed by Me2-em3 (11 bands) while Me3-em3 and Me2-em2 recorded the lowest band number (6 bands). The genotype-specific SRAP markers for the different EPN species are listed in Table 7. The highest number of SRAP specific markers was scored for S. glaseri (7 markers), then 4 markers for S. abbasi 2, followed by S. riobrave (3 markers), one marker for S. abbasi 1 while none for S. carpocapsae DD 136, S. carpocapsae all, and S. scapterisci. Devran and Baysal (2012) used 20 SRAP primer combinations to evaluate the genetic variations in M. incognita isolates. They found that the highest number of polymorphic bands was produced by Me5-em4 SRAP primer combination, while the lowest number of polymorphic bands was obtained by Me1-em1 SRAP primer combination.

The genetic variability among nematode species as revealed by SRAP analysis, M, 1 kbp plus DNA ladder, 1–7, the nematode species (1, S. abbasi 1; 2, S. abbasi 2; 3, S. carpocapsae DD 136; 4, S. carpocapsae all; 5, S. riobrave; 6, S. scapterisci; 7, S. glaseri)
The relationships among seven species of EPN
The obtained data from RAPD markers were used to calculate the phylogenetic tree which revealed the genetic relationship among different species of EPN. The cluster dendrogram showed two major clusters (Fig. 3). The first cluster was divided into two branches, the first one had S. abbasi 1, and the second branch had S. abbasi 2. The second cluster was subdivided into two branches, the first one was S. glaseri and the other branch was divided into two sub-cluster, the first contained S. carpocapsae DD 136 and the second contained S. carpocapsae all, S. riobrave, and S. scapterisci.

Phylogenetic tree of seven steinernematid species based on pooled RAPD markers
The phylogenetic tree from SRAP markers showed two major clusters as RAPD phylogenetic tree (Fig. 4). The first cluster was divided into two branches, the first one has S. abbasi 1 and the second branch had S. abbasi 2. The second cluster was subdivided into two branches, the first one is S. glaseri and the other branch was divided into two sub-clusters, the first contained S. riobrave and S. scapterisci and the second contained S. carpocapsae DD 136 and S. carpocapsae all. Therefore, the two molecular markers can be used to differentiate among the seven species of EPN.

Phylogenetic tree of seven steinernematid species based on pooled SRAP markers
Similar results were reported by Dillman et al. (2015) who sequenced and accumulated the genomes of five Steinernema spp. (S. carpocapsae, S. feltiae, S. glaseri, S. monticolum, S. scapterisci) and supposed the relationships among the five Steinernema. S. carpocapsae and S. scapterisci had the same cluster and they were more closely to S. feltiae than S. glaseri whereas in the present study, S. carpocapsae was near to S. glaseri than S. scapterisci.
Conclusion
The present study indicated that RAPD and SRAP can be successfully used as molecular markers for estimating the genetic variability and the genetic relationships among the EPNs. Specific molecular markers can be used as specific probes for identification of different species and isolates, which can be used in biological control programs.
Change history
18 April 2019
Following publication of the original article [1], the authors flagged that the titles of Table 1 and Table 2 were the wrong way around.
References
Abedian M, Talebi M, Golmohammdi HR, Sayed Tabatabaei BE (2012) Genetic diversity and population structure of mahaleb cherry (Prunus mahaleb L.) and sweet cherry (Prunus avium L.) using SRAP markers. Biochem Syst Ecol 40:112–117
Adams BJ, Fodor A, Koppenhofer HS, Stackebrandt E, Stock SP, Klein MG (2006) Biodiversity and systematics of nematode-bacterium entomopathogens. Biol Control 37:32–49
Agarwal M, Shrivastava N, Padh H (2008) Advances in molecular marker techniques and their applications in plant sciences. Plant Cell Rep 27:617–631
Budak H, Shearman RC, Parmaksiz I, Dweikat I (2004) Comparative analysis of seeded and vegetative biotype buffalo grasses based on phylogenetic relationship using ISSRs, SSRs, RAPDs, and SRAPs. Theor Appl Genet 109:280–288
Devran Z, Baysal O (2012) Genetic characterization of Meloidogyne incognita isolates from Turkey using sequence-related amplified polymorphism (SRAP). Biologia 67:535–539
Dillman AR, Macchietto M, Porter CF, Rogers A, Williams B, Antoshechkin I, Lee M, Goodwin Z, Lu X, Lewis EE, Goodrich-Blair H, Stock SP, Adams BJ, Sternberg PW, Mortazavi A (2015) Comparative genomics of Steinernema reveals deeply conserved gene regulatory networks. Genome Biol 16:20
Dutky SR, Thompson JV, Cantwel GE (1964) A technique for the mass propagation of the DD-136 nematode. J insect Pathol 6:417–422
Gardner SL, Stock SP, kava HK (1994) A new species of Heterorhabditis from the Hawaiian islands. J Parasitol 80:100–109
Gasser RB, Hu M, Chilton NB, Campbell BE, Jex AJ, Otranto D, Cafarchia C, Beveridge I, Zhu X (2006) Single-strand conformation polymorphism (SSCP) for the analysis of genetic variation. Nat Protoc 1:3121–3128
Hashmi G, Gaugler R (1997) Genetic diversity in insect parasitic nematodes (Rhabditida: Heterorhab-ditidae). J Nematol 29:582–584
Hashmi G, Gaugler R (1998) Genetic diversity in insect-parasitic nematodes (Rhabditida: Heterorhabditidae). J Invertebrate Pathol 72:185–189
Hashmi G, Itamar G, Gaugler R (1996) Molecular comparisons of entomopathogenie nematodes using randomly amplified polymorphie DNA (RAPD) markers. Fund Appl Nematol 19(4):399–406
Ibrahim SAM (2009) Genetic diversity and phylogenetic relationships of some entomopathogenic nematode species (Steinernematidae and Heterorhabditidae). Int J Nematol 19(2):144–150
Kary NE, Niknam G, Griffin CT, Mohammadi SA, Moghaddam MA (2009) Survey of entomopathogenic nematodes of the families Steinernematidae and Heterorhabditidae (Nematoda: Rhabditida) in the north-west of Iran. Nematol 11:107–116
Laznik Z, Trdan S (2014) The influence of insecticides on the viability of entomopathogenic nematodes (Rhabditida: Steinernematidae and Heterorhabditidae) under laboratory conditions. Pest Manag Sci 70:784–789
Li G, Quiros CF (2001) Sequence-related amplified polymorphism (SRAP), a new marker system based on a simple PCR reaction: its application to mapping and gene tagging in Brassica. Theor Appl Genet 103(2–3):455–461
Li XY, Li J, Zhao ZJ, Yang F, Fu QW, Liu HS, Wang DD, Yang YC, Wang RY (2014) Sequence-related amplified polymorphism (SRAP) for studying genetic diversity and population structure of plants and other living organisms. J Anim Plant Sci 24(5):1478–1486
Liu J, Berry RE (1996) Steinernema oregonensis n. sp. (Rhabditida: Steinernematidae) from Oregon, USA. Fund Appl Nematol 19:375–380
Moghaieb REA, Ibrahim SAM, Saneoka H, Fujita K (2004) Molecular characterization of heat tolerance in two isolates (HTS1 and HTS2) derived from the entomopathogenic nematode strain ISK-2 (Heterorhabditis species). Int J Nematol 14(1):66–72
Mutlu N, Boyaci FH, Göçmen MM, Abak K (2008) Development of SRAP, SRAP-RGA, RAPD and SCAR markers linked with a Fusarium wilt resistance gene in eggplant. Theor Appl Genet 117:1303–1312
Padmanaban K, Karthik RR, Razia M, Chellapandi P, Sivaramakrishnan S (2014) Genetic diversity of entomopathogenic nematodes in non-agricultural ecosystem revealed with PCR-RAPD markers. Int J Nematol 24(2):1–8
Shapiro DI, Glazer I, Segal D (1997) Genetic diversity in wild and laboratory population of Heterorhabditis bacteriophora as determined by RAPD–PCR analysis. Fund Appl Nematol 20:581–585
Stock S, Strong D, Gardner S (1996) Identification of Heterorhabditis (Nematoda: Heterorhabditidae) from California with a new species isolated from the larvae of the ghost moth Hepialis californicus (Lepidoptera: Hepialidae) from the Bodega Bay natural reserve. Fund Appl Nematol 19:585–592
Zhao GH, Li J, Zou FC, Mo XH, Yuan ZG, Lin RQ, Weng YB, Zhu XQ (2009) ISSR, an effective molecular approach for studying genetic variability among Schistosoma japonicum isolates from different provinces in mainland China. Infect Genet Evol 9:903–907
Acknowledgements
We would like to thank Dr. Reda E.A. Moghaieb (Genetic Engineering Research Center, Faculty of Agriculture, Cairo University, Egypt) for helping in statistical analysis, critical reading of the manuscript.
Funding
Not applicable
Availability of data and materials
All datasets are presented in the main manuscript.
Author information
Authors and Affiliations
Contributions
AEAA participated in the experimental design, practical work, and manuscript writing. KE participated in the experimental design and practical work and coordinated in manuscript writing. IS participated in discussion and provided intellectual support to this research. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional information
The original version of this article was revised to correct the titles of Tables 1 and 2, which had been erroneously swapped around in the originally published article.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Abd ElAzim, A.M., Khashaba, E.H.K. & Ibrahim, S.A.M. Genetic polymorphism among seven entomopathogenic nematode species (Steinernematidae) revealed by RAPD and SRAP analyses. Egypt J Biol Pest Control 29, 17 (2019). https://doi.org/10.1186/s41938-019-0114-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41938-019-0114-z