
Abstract
Background
Next-generation sequencing (NGS) based assay for finding an actionable driver in non-small-cell lung cancer is a less used modality in clinical practice. With a long list of actionable targets, limited tissue, arduous single-gene assays, the alternative of NGS for broad testing in one experiment looks attractive. We report here our experience with NGS for biomarker testing in hundred advanced lung cancer patients.
Methods
Predictive biomarker testing was performed using the Ion AmpliSeq™ Cancer Hotspot Panel V2 (30 tumors) and Oncomine™ Solid Tumor DNA and Oncomine™ Solid Tumor Fusion Transcript kit (70 tumors) on Ion-Torrent sequencing platform.
Results
One-seventeen distinct aberrations were detected across 29 genes in eighty-six tumors. The most commonly mutated genes were TP53 (43% cases), EGFR (23% cases) and KRAS (17% cases). Thirty-four patients presented an actionable genetic variant for which targeted therapy is presently available, and fifty-two cases harbored non-actionable variants with the possibility of recruitment in clinical trials. NGS results were validated by individual tests for detecting EGFR mutation, ALK1 rearrangement, ROS1 fusion, and c-MET amplification. Compared to single test, NGS exhibited good agreement for detecting EGFR mutations and ALK1 fusion (sensitivity- 88.89%, specificity- 100%, Kappa-score 0.92 and sensitivity- 80%, specificity- 100%, Kappa-score 0.88; respectively). Further, the response of patients harboring tyrosine kinase inhibitor (TKI) sensitizing EGFR mutations was assessed. The progression-free-survival of EGFR positive patients on TKI therapy, harboring a concomitant mutation in PIK3CA-mTOR and/or RAS-RAF-MAPK pathway gene and/or TP53 gene was inferior to those with sole-sensitizing EGFR mutation (2 months vs. 9.5 months, P = 0.015).
Conclusions
This is the first study from South Asia looking into the analytical validity of NGS and describing the mutational landscape of lung cancer patients to study the impact of co-mutations on cancer biology and treatment outcome. Our study demonstrates the clinical utility of NGS testing for identifying actionable variants and making treatment decisions in advanced lung cancer.
Similar content being viewed by others

Background
Cancer is recognized as a genetic disorder. Genetic alterations in the lung adenocarcinoma (LUAD) have been well documented and are observed in more than 60% of the cases. Identifying a driver mutation in one of the several oncogenes like EGFR, ALK, ROS1, BRAF, KRAS, AKT1, HER2, MEK1, MET, NRAS, PIK3CA and RET can have therapeutic bearings in LUAD [1, 2]. Driver mutation-based targeted therapies, wherever possible, improve the median overall survival of patients with metastatic LUAD by at least one year [3]. Single-gene assays to identify actionable mutations is the current standard of care in advanced LUAD. However, for identifying actionable driver mutations beyond EGFR sensitizing mutations, ALK1 and ROS1 fusion rearrangements, next-generation sequencing (NGS) is the most practical option, given the limited availability of biopsy material and arduous single-gene assays. NGS based genetic profiling of advanced solid tumors is a relatively new technique and allows comprehensive search for predictive biomarkers in a resource and tissue proficient manner [4,5,6,7,8]. Moreover, the broader molecular profile by NGS allows evaluation of the variants of potential clinical significance (Tier II genetic alterations) leading to many additional patients drawing benefits of targeted therapy [9].
To establish the theoretical advantages of NGS over single-gene assay in the real world, we undertook biomarker testing for advanced non-small-cell lung cancer (NSCLC) patients by NGS and compared the results with single-gene assays to determine its accuracy, reliability, and benefits in understanding the cancer biology in relation to the effect of co-mutations on treatment results and survival statistics.
Methods
The study was approved by our ‘Institutional Review Board’ (RGCIRC/ IRB/ 277/ 2019) and conducted in accordance with the Declaration of Helsinki. Clinical characteristics, treatment details, and outcome were curated from the electronic medical record of the institute.
The study is single institutional; the time period spans from January 2015 to December 2018. The cases included were consecutive advanced lung cancer patients diagnosed as NSCLC- adenocarcinoma, NSCLC- not otherwise specified and a few squamous cell carcinoma patients selected by physician based on clinical features with a high likelihood of finding a driver mutation. Predictive biomarker testing by NGS and single gene tests were performed at the time of diagnosis.
A total of hundred patients were tested on NGS using ‘Ion AmpliSeq™ Cancer Hotspot Panel V2’ (4475346, Thermo Fisher Scientific) for the first thirty cases, and ‘Oncomine™ Solid Tumor DNA and Oncomine™ Solid Tumor Fusion Transcript Kit’ (A26761, A26762, Thermo Fisher Scientific) for the later seventy cases. The genes interrogated by Ion AmpliSeq™ Cancer Hotspot Panel v2 (hotspot regions of 50 oncogenes/ tumor suppressor genes) and the Oncomine™ Solid Tumor DNA & Oncomine™ Solid Tumor Fusion Transcript kits (DNA somatic variants in 22 key solid tumor genes and RNA fusion transcript in ALK, RET, ROS1 and NTRK1) has been shown in Additional file 1.
Isolation of nucleic acid
Formalin-fixed and paraffin-embedded (FFPE) tumor biopsies were reviewed by AM. Slides with ~ 10% or more tumor area were selected or obtained after macrodissection. Genomic DNA was isolated from FFPE sections (5x, 10 μm) using the Promega ReliaPrep™ FFPE gDNA Miniprep System (A2352, USA) and RNA was extracted using the Promega ReliaPrep™ FFPE Total RNA Miniprep System (Z1002, USA). Nucleic acid was quantitated by Qubit® 3.0 Fluorometer (Invitrogen Life Technologies). cDNA was generated from 10 ng of RNA using the SuperScript™ VILO™ cDNA synthesis kit (11754050, USA). The protocol followed was according to the vendor’s insert.
Library preparation and ion-torrent based NGS
Amplicon library was prepared using 10 ng of DNA/ cDNA using the respective targeted panel. Primers were partially digested and the amplicons were phosphorylated with the FuPa reagent. Sample barcoding was performed using Ion Dx barcodes and the samples were adaptor-ligated. The amplified product was purified and the sequencing library was prepared with Ion AmpliSeq™ Library Kit Plus (A35907, Thermo Fisher Scientific). Consequently, emulsion PCR was performed using Ion PI™ Hi-Q™ One Touch2 (OT2) 200 template kit (A26434) and the library was enriched on Ion One Touch™ ES instrument. Sequencing was performed employing Ion PGM™ Hi-Q™ Sequencing kit (A25592) on Ion 318™ Chip v2 (8–10 samples on a single chip for each sequencing run), on the Ion Personal Genome Machine™ (PGM™) System (Thermo Fisher Scientific). The protocol followed at each step was in line with manufacturers’ instructions without any modifications.
NGS data analysis
Sequencing data were checked for quality metrics using the Torrent Suite version 5.0.2 (Thermo Fisher Scientific). Somatic variants and fusion transcripts were called by Ion Reporter using the specific BED files employing appropriate filters. Integrative Genomic Viewer (IGV) version 2.3 (or higher) was used to verify the variants called and to identify short reads with potential mispriming events. The Oncomine™ Knowledgebase Reporter Software (Thermo Fisher Scientific) was used for the final report generation, based on the sequence alignment with the reference genome hg 19. The threshold of the mutation frequency was 2% at a median coverage depth of >1000X. Variants of unknown significance were checked on the VarSome search engine which allows access to publications, ClinVar and all in silico prediction tools on one single site [10, 11]. Further, the OncoPrint heat map and mutation plots were generated using the online customized tools OncoPrinter and MutationMapper, respectively, at cBioPortal for Cancer Genomics [12, 13]. All the pathogenic mutations were checked in the NCBI and COSMIC databases [14, 15]. TP53 mutations were compared and analyzed in the IARC TP53 database (version R19) [16].
Single tests
For determination of analytical and clinical validity of NGS vis-à-vis reference method of single-gene assay, four single-gene analyses for sensitizing mutations in the EGFR gene, rearrangement of ALK1 and ROS1 and amplification of c-MET were performed on all cases where adequate tissue or cytology material was available.
EGFR mutation analysis
Mutational analysis for EGFR was done using Qiagen EGFR Therascreen® RGQ PCR Kit (870111). Five sections of 4 μm each were collected in an Eppendorf tube with manual macro-dissection to enrich tumor fraction wherever necessary. DNA was extracted using Qiagen DNeasy blood and tissue kit (69504). Multiplex Real-Time PCR was carried out on Rotor-Gene Q thermal cycler (Qiagen) in 8 tubes along with positive and no-template controls. The interpretation was done as per the vendor’s insert.
Immunohistochemistry for ALK1 protein
ALK1 protein was tested by immunohistochemistry (IHC) using anti-ALK (D5F3) rabbit monoclonal primary antibody with other proprietary components of the VENTANA ALK (D5F3) CDx assay (790–4796, Roche) on Ventana BenchMark XT automated slide stainer (using Ventana’s OptiView DAB IHC Detection kit and OptiView Amplification kit); performed according to the manufacturer’s recommendations.
Detection of ROS1 rearrangement and c-MET gene amplification by fluorescence in situ hybridization
Fluorescence in situ hybridization (FISH) was performed on FFPE lung tissue sections of 4–5 μm, placed on positively charged slides. The specimens used for this study were hybridized using break-apart probe set (ZytoLight® SPEC ROS1 Dual Color Break Apart Probe, ZytoVision, GmbH, Germany), according to the manufacturer’s instructions. FISH measurements were performed using fluorescence microscope Leica DM6000B (Leica, Japan). The hybridized sections were examined under 100x magnification for break-apart signals. A distance of more than 1 signal diameter between red and green signals was considered positive. Lesser than 5 split signals were reported negative and > 25 split signals were considered positive on a count of 50 cells. In the case of 6–24 split signals, a second operator repeated the count. An average of ≥15% signals was considered positive.
c-MET in situ hybridization was done as per the manufacturer’s protocol (ZytoLight directly labeled LSI MET DNA probe; green and CEN-7 probe; orange). A centromeric 7 probe to MET signal ratio > 2.5 with an average number of ≥5 MET signals was considered positive.
Targeted therapy response in EGFR mutated tumors
Therapeutic decision was made according to the NGS test results. Based on the NGS profile, the EGFR mutated patients were separated into two groups as those with (i) isolated EGFR mutations and (ii) compound mutations with a concurrent mutation in PIK3CA – mTOR pathway/ MAPK pathway and/ or TP53 mutation. The response of the patients put on small molecule Tyrosine Kinase Inhibitor (TKI) was assessed. Those patients who showed complete/ partial response were classified as responders. While those with stable/ progressive disease were grouped as non-responders to the treatment. The radiological response was evaluated by the treating physician according to the Response Evaluation Criteria In Solid Tumors (RECIST). The interval between the computed tomography scan was once in 3–4 months/ a smaller interval as directed by the treating physician.
Statistical analysis
Descriptive statistics were used to summarize data. Association between the EGFR mutation status and gender/ smoking history was analyzed using Pearson’s chi-squared test. The diagnostic test evaluation was performed on MedCalc for Windows, version 15.8 (MedCalc Software bvba, Ostend, Belgium). The sensitivity, specificity, and positive and negative predictive values (NGS versus single tests) were computed by taking the single-gene assay results as a reference. Further, the concordance of the two techniques was measured by Cohen’s Kappa statistics.
The Progression-Free Survival (PFS) was calculated from the date of the start of TKI till the date of radiological progression/ death. Kaplan-Meier survival curves for the single and compound EGFR gene mutation groups were plotted and compared by the log-rank test. The limit of statistical significance was set as 0.05 (5% level). Statistical analyses were performed using the SPSS version 23.0 software package (IBM Corp, Armonk, NY).
Results
Patient characteristics
The baseline characteristics of the 100 advanced NSCLC patients tested on the multigene panel have been presented in Table 1. Majority tumors (98%) were adenocarcinoma and two (2%) were squamous cell carcinoma.
Distribution of the oncogenic driver mutations in the cohort
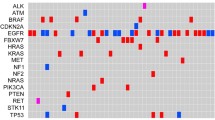
Targeted sequencing identified pathogenic alterations in 28 different genes (Additional file 1). Moreover, copy number change in the gene CDK4 (not included in the panel) was called for in a single case. The gene fusions for ALK1, RET, ROS1 and NTRK1 were tested in 70 tumors. The variant data summary as visualized on the OncoPrint heat map has been shown in Fig. 1. Most patients (44 cases) presented single-gene mutation, 27 cases presented mutations in two genes, 10 cases harbored mutations in three genes and a lesser number (5 cases) presented more than three mutations. Among these, the commonest genetic alteration was in the TP53 gene (43% cases), followed by EGFR (23%) and KRAS (17%). The frequency of mutation in other genes ranged from 1 to 7% (Fig. 1). The mutational plots for TP53, EGFR and KRAS genes have been displayed in Fig. 2 (A-C). Most of the pathogenic TP53 mutations localized to the DNA-binding domain. In total 40 distinct TP53 mutations were detected, among them the p.P72R was the most frequent variant (n = 10). Missense TP53 mutations were common (35 of the 40 distinct TP53 variants, 87.5%), while 2 microindels (p.G293fs and p.P301fs) leading to a frameshift (5%) and three protein-truncating nonsense mutations p.R213*, p.R306* and p.E349* (7.5%) were observed (Fig. 2A). The mutations were further analyzed on the IARC TP53 database (Additional file 2). All the TP53 mutations identified in the cohort have been previously reported. One significant observation that emerged from the IARC TP53 database was that 32 of the 35 missense TP53 mutations were pathogenic as per the SIFT predictions (Additional file 2E).

OncoPrint showing the distribution of genomic alterations in 29 genes and 100 lung cancer cases. Note: For cases 1–30 the IonAmpliSeq™ Cancer Hotspot Panel V2 was used, and in cases 31–100 the Oncomine™ Solid Tumor DNA and Oncomine™ Solid Tumor Fusion Transcript kit was employed. Frequency (%) for the genes APC, ATM, FLT3, GNAQ, IDH2, JAK3, KDR, KIT, SMARCB1 and SMO have been calculated in 30 patients; and for fusion transcripts in ALK, RET and ROS1 has been calculated among the 70 tested cases. The frequency for all other genes, common to both the panels has been calculated in 100 cases. The red, green and blue asterisk symbol (*) indicates patients with ≥3, 2 and 1 pathogenic mutation(s), respectively. No mutation was detected in 14 cases

Lollipop plots depicting the distribution of (A) TP53 (B) EGFR (C) KRAS mutations detected on NGS in the study group (N = 100, carcinoma lung subjects). Please note: The four uncommon EGFR gene mutations (p.D770delinsES, p.D770_771insG, p.A767_S768insSVD, p.P772_H773insHV) have been depicted as ‘U’ in the panel B. (GenBank Reference TP53: NM_000546, EGFR: NM_005228, KRAS: NM_033360)
EGFR gene alterations were detected in 23/100 cases. Among the adenocarcinoma patients, the EGFR mutations were more frequent in females than in the males (32.6% versus 15.4%, P = 0.045) and in never-smokers than the ever-smokers (32.3% versus 9.5%, P = 0.041). Deletion in exon 19 was the commonest mutation (Fig. 2B). A coexistent p.T790M mutation was observed in three tumors. Four uncommon EGFR Exon 20 insertion mutations were detected (p.D770delinsES, p.D770_771insG, p.A767_S768insSVD and p.P772_H773insHV). Of interest was a single subject who showed a gamut of EGFR alterations that included inframe exon 19 deletion, EvIII fusion and copy number gain.
Somatic mutation in the KRAS gene occurred in 17/100 subjects. These mutations were missense substitutions that changed the amino acid glycine in codon 12/13 (Fig. 2C).
In total NGS detected 117 distinct pathogenic alterations in 29 different cancer-driver genes. To correlate these aberrations clinically, they were categorized into three groups (i) Tier I: driver mutations that are actionable by Food and Drug Administration (FDA) approved targeted therapies (EGFR, ALK1 fusions, ROS1 fusions and BRAF (p.V600E) (ii) Tier II: alterations in well-known cancer oncogenes, actionable by targeted agents not-yet-approved by FDA (ERBB2, RET and MET amplification, and MET Exon 14 skipping mutation) (iii) Tier III: Clinically significant, non-actionable variants (all other genes).
Clinically actionable and non-actionable variants detected by sequencing
The alterations observed related to approved/ emerging treatments have been enlisted in Additional file 3. In total, fourteen different variants/ mutation subtypes were seen in Tier I genes (12%) and six in Tier II genes (5%). Ninety-seven alterations (83%) were detected in Tier III genes (Fig. 3, Additional file 3). In terms of patient population showing Tier I and Tier II gene mutation type, 29 cases (29%) and 5 cases (5%), respectively, were identified. There were 14 cases (14%) without any somatic mutation in the tested genes. While 52 cases (52%) had non-actionable genomic alterations (Tier III) (Fig. 3). Two novel Tier III variants in the genes FGFR3 (p.G90del) and IDH2 (p.T138A) were identified which have not been reported previously (Additional file 3). Twenty-two patients harbored more than one mutation belonging to Tiers I & II/ I & III/ II & III / I, II & III.

Dough-nut plot showing the percentage of patients with clinically actionable (Tier I and Tier II) and non-actionable (Tier III) genetic alterations detected by NGS in the study group (N = 100, Lung cancer patients)
Comparison of NGS and single testing methods for detecting genomic alterations in EGFR, ALK1, ROS1 and c-MET
We next validated the NGS results for four genes for which single assay as mentioned in the methods section were performed to study the concordance and analytical validity of NGS. Due to limited tissue availability, single tests were not done in all the samples. Also, NGS test for identifying rearranged ALK1 and ROS1 were performed in 70 cases (those tested on the Oncomine™ Solid Tumor Fusion panel). Therefore, in total 75, 49, 43 and 62 cases each have been compared for EGFR mutation, ALK1 rearrangement, ROS1 fusion and c-MET amplification, respectively, for the single test and NGS outcome (Table 2).
Among the seventy-five EGFR gene mutation tested patients, results were concordant in all except for two cases. Discordance was observed in two subjects harboring p.L858R mutation. Compared to multiplexed real-time PCR, the sensitivity and specificity of the NGS assay was 88.89 and 100%, respectively. Both methods achieved almost perfect agreement (Kappa-score = 0.92) (Table 3). Similarly, for the detection of ALK1 rearrangement, both NGS and IHC methods positively confirmed 4 cases, whereas the result was discordant for a single patient. The sensitivity and specificity estimates were 80 and 100%, respectively (Kappa-score = 0.88). With respect to the detection of ROS1 fusion and c-MET expression, the specificity of both diagnostic methods was high, but sensitivity was not determined. The single ROS1 fusion variant detected by NGS went undetected on FISH. Also, results varied for c-MET amplification. NGS missed the four c-MET amplified cases that were detected on FISH (Table 3).
Response to targeted therapy in patients with single and compound EGFR gene mutations
To determine the impact of co-mutations on treatment response, we examined the PFS of the twenty-three cases harboring isolated EGFR mutations, or EGFR mutations along with mutations in genes involved in the RAS/RAF/MEK/ERK/MAPK or PIK3CA/AKT/mTOR pathway and/ or concomitant TP53 mutation (Additional file 4). The response was not evaluated in three cases (two external outpatient cases and one terminally ill patient). Also, the four cases with uncommon EGFR gene mutations were excluded from the PFS analysis, as these patients were not treated by TKI inhibitors.
TP53 gene mutations were observed in 8/23 (34%) subjects. While 6/23 (26%) subjects each showed co-mutations of EGFR with KRAS-BRAF-MAPK pathway genes or with PIK3CA-mTOR pathway genes. In total, 16 patients received TKI therapy. Eight patients (50%) harbored co-mutations, and showed a significantly shorter PFS than those with single EGFR gene mutation [median PFS = 2 months, 95% CI (0.00–5.46) versus 9.5 months, 95% CI (0.52–18.5), respectively; PLog Rank = 0.015] [Fig. 4 (A-C)].

(a) Kaplan-Meier survival curve showing Progression-Free Survival (PFS) in patients with isolated single and compound EGFR mutations (n = 16). (b) Pathway specific co-mutations (encircled) as observed in the compound EGFR mutant group. (c) OncoPrint presenting the summary of the twenty-three EGFR mutated tumors for the single [1,2,3,4,5,6,7,8,9,10,11] and compound [12,13,14,15,16,17,18,19,20,21,22,23] mutation groups and the patients’ response to TKI therapy. The response could not evaluated in the cases 9, 10, 11, 20, 21, 22 and 23 as they were outpatients/ not treated by TKI. A: Afatinib, E: Erlotinib, G: Gefitinib, O: Osimertinib
Discussion
Predictive biomarker identification with cognate targeted therapy has improved the treatment outcomes in NSCLC. The standard single-gene assays are demanding in terms of both tissue and time. Next-generation sequencing techniques interrogate several cancer-driver-gene alterations, thereby providing a mutational portrait even in those tumors which have a low tumor fraction. Short turnaround time can be another advantage if the volume of tests available is optimal for chip/ flow cell usage. Despite these benefits, molecular testing requires performance characteristics of the NGS techniques acceptable in terms of analytical validity, clinical validity and clinical utility vis-a-vis the single-gene assays. This is the first study from South Asia looking into the analytical validity of NGS and describing the mutational landscape of lung cancer patients to study the impact of co-mutations on cancer biology and treatment outcome. NGS technique was applied to FFPE tumor blocks of hundred lung cancer subjects. The most commonly mutated gene was TP53 (43%). This frequency is similar to a study by Tsoulos N. et al. on 502 NSCLC patients but is lower to ‘The Cancer Genome Atlas’ (TCGA) data (51.8%) [17,18,19,20]. About 88% of the TP53 mutations identified in our study were missense mutations and yet were pathogenic as per the IARC database. The high rate of missense pathogenic mutations is unique to TP53 where unlike other genes the frameshift and nonsense mutations are infrequently pathogenic. Also, we observed a high prevalence TP53 alteration ‘p.P72R’ (10 cases, 19.2%) at an allele frequency of approximately 50% suggesting germline status of this variant. Shi et al. reported a high allele frequency of the p.P72R in Eastern Asia [21]. This SNP is known as PEX4 (pleomorphism in exon 4), has been reported at reasonably high rates worldwide though its prevalence in the Indian population is unknown because of a lack of an available database. The residue 72 in the TP53 protein is not conserved residue to severely hamper protein structure and function. Besides, proline is physiochemically not too different from arginine [22]; yet this SNP has been shown by Wang and colleagues in their large meta-analysis evaluating “the strength of evidence of published candidate-genes association studies in lung cancer”, to have a significant association with lung cancer susceptibility [23]. This SNP has been widely researched [22, 24, 25] and has been linked with a small and definitive risk of several sporadic cancers due to reduced functional efficiency of p53 protein coded on this SNP.
Around 23% of the cases presented aberrations in the EGFR gene. This frequency matches a large cohort study from India by Chougule et al. [26]. Also, consistent with previous studies, the EGFR gene mutations were more frequent in women and in non-smokers in the LUAD patients [27]. Four rare EGFR Exon 20 insertion mutations were detected by NGS, and it’s rate in EGFR mutated patients 17.4% (4/23) was comparable to a recent study from India that has reported the rate as 18.07% (15/83) [28]. Also, similar to previous studies all the KRAS mutations detected in our study were located on Exon 2 [29]. A comparison of the prevalence of frequent driver gene alterations observed in this study with the data from the TCGA database for LUAD and that of a recent study in East-Asian patients has been presented in Additional file 5, where a significant difference in the prevalence of EGFR and KRAS mutations were observed.
Among the NGS tested cases, 34% (34/100) patients presented an actionable genomic variant according to the NCCN guidelines [30, 31]. Among them, the results of NGS and single-gene assays for identifying EGFR mutations and ALK1 fusion demonstrated good agreement, while the results were more discordant for MET and ROS1 genes. Copy number gain false negativity is likely due to failure to get adequate amplicons during library preparation compared to normalization genes. FISH assay for ROS1 is fraught with technical inconsistencies like the inability of probes to hybridize and difficult interpretations and may have contributed to false-negative results on the FISH assay. Significantly, patients detected by NGS to have ROS1 fusions which were negative by FISH responded to Crizotinib and prove the superiority of the NGS platform to detect this biomarker. In a retrospective study by Legras et al., TaqMan probes and NGS were compared for their ability to detect EGFR and KRAS mutations, and NGS mutation profiles were studied on a large series of NSCLC patients (n = 1343) [7]. The results showed a high concordance of the two techniques, with a Kappa-score for the detection of EGFR gene mutation as 0.99 (95% CI: 0.97–1.00) [7]. The Kappa-value observed in the present study 0.92 (95% CI: 0.82–1.00) agrees with the aforementioned study. Study by Tsoulos et al. showed 100% concordance between high resolution melting curve analysis and NGS for detecting mutations in EGFR gene (exons 18, 19, 20 and 21), KRAS (exon 2, 3 and 4), and NRAS (exons 11 and 15), wherein NGS techniques demonstrated enhanced sensitivity [17]. Other studies by de Leng et al. and Jing et al. also shows good agreement of NGS and single-gene assays for detecting driver-gene mutations in NSCLC, exhibiting high sensitivity and specificity, suggesting the possibility of routine use of NGS assays to guide clinical decisions [29, 32]. In the latter study, the performance of NGS and digital droplet PCR, a technique with high analytical sensitivity (< 1%), were comparable for detecting EGFR mutations.
Previous studies have shown that compound EGFR mutations and concurrent genomic alterations with EGFR active mutations are associated with inferior clinical outcome in EGFR mutated LUAD patients [20, 33, 34]. The study by Barnet et al. observed significantly shorter PFS in the NSCLC patients harboring compound mutations (dual EGFR/ PIK3CA mutations) than the single EGFR mutated cases [34]. Some studies indicate that TP53 effects P13K/AKT and ERK pathways and mutated TP53 fails to induce apoptosis in response to TKI [35, 36]. A similar observation was made in the current study also with 5/16 EGFR positive patients with a co-mutation in the TP53 gene showed an inferior response to TKI therapy.
Patients with sole EGFR sensitizing mutations showed significantly longer PFS to those who showed additional mutations in any one of the three alluded pathways. Future studies dedicated to the compound gene mutation group of the patients including the uncommon mutation as a group are required to further establish the usage of current targeted therapy in such patients. Also, the imaging performed early in non-responders could lead to a shorter response interval in the group. A prospective study comparing cohorts with isolated sensitizing EGFR mutations against those with dual or several concurrent mutations with a predetermined interval for response evaluation will bring further clarity on this issue.
Lastly, it must be admitted that though NGS allow multiplexing and offer broad coverage for oncogenes/ tumor suppressor genes, it is associated with variable error rate (0.1–15%) that is often encountered for short reads obtained from FFPE samples [37,38,39]. This necessitates a larger prospective study with objective response rates and survival being the study endpoint apart from analytical validity of the NGS.
Conclusions
Our study demonstrates the value of NGS in biomarker testing for advanced/ metastatic NSCLC. The high concordance of NGS and single-gene assay results establish the analytical validity of NGS. Further, NGS allows the identification of additional patients with clinically actionable variants from Tier II who can potentially benefit from targeted therapy. The broader genomic picture promotes the understanding of mutational interactions in determining the response to targeted therapies. The smaller number of cases and limited targeted panel instead of a panel with all significantly mutated genes capable of finding all high confidence drivers is the limitation of this study along with limited follow-up and survival data. The strength of this work is the prospective nature of the study and the use of all FDA approved test methodologies (wherever available) for single-gene assays.
Availability of data and materials
The data generated and analysed in the study are included in the published article/ as supplementary information. Additional datasets will be made available from the corresponding author on a reasonable request.
Abbreviations
- EGFR:
-
Epidermal Growth Factor Receptor
- FDA:
-
Food and Drug Administration
- FFPE:
-
Formalin-fixed and paraffin-embedded
- FISH:
-
Fluorescence in situ hybridization
- IHC:
-
Immunohistochemistry
- LUAD:
-
Lung adenocarcinoma
- NGS:
-
Next-generation sequencing
- NSCLC:
-
Non-small-cell lung cancer
- PFS:
-
Progression-free survival
- RECIST:
-
Response Evaluation Criteria In Solid Tumors
- SNP:
-
Single nucleotide polymorphism
- TKI:
-
Tyrosine kinase inhibitor
- TCGA:
-
The Cancer Genome Atlas
References
Thunnissen E, van der Oord K, den Bakker M. Prognostic and predictive biomarkers in lung cancer. A review. Virchows Arch. 2014;464:347–58.
Lovly C, L. Horn, W. Pao. Molecular profiling of lung cancer. My Cancer Genome. 2018. https://www.mycancergenome.org/content/disease/lung-cancer/.
Kris MG, Johnson BE, Berry LD, Kwiatkowski DJ, Iafrate AJ, Wistuba II, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. Jama. 2014;311:1998–2006.
Metzker ML. Sequencing technologies - the next generation. Nat rev Genet. 2010;11:31–46.
Diaz Z, Aguilar-Mahecha A, Paquet ER, Basik M, Orain M, Camlioglu E, et al. Next-generation biobanking of metastases to enable multidimensional molecular profiling in personalized medicine. Mod Pathol. 2013;26:1413–24.
Yamamoto G, Kikuchi M, Kobayashi S, Arai Y, Fujiyoshi K, Wakatsuki T, et al. Routine genetic testing of lung cancer specimens derived from surgery, bronchoscopy and fluid aspiration by next generation sequencing. Int J Oncol. 2017;50:1579–89.
Legras A, Barritault M, Tallet A, Fabre E, Guyard A, Rance B, et al. Validity of targeted next-generation sequencing in routine care for identifying clinically relevant molecular profiles in non-small-cell lung cancer: results of a 2-year experience on 1343 samples. J Mol Diagn. 2018;20:550–64.
Garinet S, Laurent-Puig P, Blons H, Oudart JB. Current and future molecular testing in NSCLC, what can we expect from new sequencing technologies? J Clin Med. 2018. https://doi.org/10.3390/jcm7060144.
Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. 2017;19:4–23.
Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Albarca Aguilera M, Meyer R, et al. VarSome: The Human Genomic Variant Search Engine. Bioinformatics. 2018. https://doi.org/10.1093/bioinformatics/bty897.
Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46:D1062–D7.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1.
Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018;46:D8-D13.
Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017;45:D777–D83.
Bouaoun L, Sonkin D, Ardin M, Hollstein M, Byrnes G, Zavadil J, et al. TP53 variations in human cancers: new lessons from the IARC TP53 database and genomics data. Hum Mutat. 2016;37:865–76.
Tsoulos N, Papadopoulou E, Metaxa-Mariatou V, Tsaousis G, Efstathiadou C, Tounta G, et al. Tumor molecular profiling of NSCLC patients using next generation sequencing. Oncol Rep. 2017;38:3419–29.
Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–9.
Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;543–50.
Sato S, Nagahashi M, Koike T, Ichikawa H, Shimada Y, Watanabe S, et al. Impact of concurrent genomic alterations detected by comprehensive genomic sequencing on clinical outcomes in east-Asian patients with EGFR-mutated lung adenocarcinoma. Sci Rep. 2018;8:1005.
Shi H, Tan SJ, Zhong H, Hu W, Levine A, Xiao CJ, et al. Winter temperature and UV are tightly linked to genetic changes in the p53 tumor suppressor pathway in eastern Asia. Am J Hum Genet. 2009;84:534–41.
Dumont P, Leu JI, Della Pietra AC, George DL, Murphy M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet. 2003;33:357–65.
Wang J, Liu Q, Yuan S, Xie W, Liu Y, Xiang Y, et al. Genetic predisposition to lung cancer: comprehensive literature integration, meta-analysis, and multiple evidence assessment of candidate-gene association studies. Sci Rep. 2017;7:8371.
Thomas M, Kalita A, Labrecque S, Pim D, Banks L, Matlashewski G. Two polymorphic variants of wild-type p53 differ biochemically and biologically. Mol Cell Biol. 1999;19:1092–100.
Whibley C, Pharoah PD, Hollstein M. p53 polymorphisms: cancer implications. Nat Rev Cancer. 2009;9:95–107.
Chougule A, Prabhash K, Noronha V, Joshi A, Thavamani A, Chandrani P, et al. Frequency of EGFR mutations in 907 lung adenocarcioma patients of Indian ethnicity. PLoS One. 2013;8:e76164.
Doval D, Prabhash K, Patil S, Chaturvedi H, Goswami C, Vaid A, et al. Clinical and epidemiological study of EGFR mutations and EML4-ALK fusion genes among Indian patients with adenocarcinoma of the lung. Onco Targets Ther. 2015;8:117–23.
Kate S, Chougule A, Joshi A, Noronha V, Patil V, Dusane R, et al. Outcome of uncommon EGFR mutation positive newly diagnosed advanced non-small cell lung cancer patients: a single center retrospective analysis. Lung Cancer (Auckl). 2019;10:1–10.
Jing C, Mao X, Wang Z, Sun K, Ma R, Wu J, et al. Next-generation sequencing-based detection of EGFR, KRAS, BRAF, NRAS, PIK3CA, Her2 and TP53 mutations in patients with nonsmall cell lung cancer. Mol Med Rep. 2018;18:2191–7.
Lindeman NI, Cagle PT, Aisner DL, Arcila ME, Beasley MB, Bernicker EH, et al. Updated molecular testing guideline for the selection of lung cancer patients for treatment with targeted tyrosine kinase inhibitors: guideline from the College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology. Arch Pathol Lab Med. 2018;142:321–46.
Ettinger DS, Aisner DL, Wood DE, Akerley W, Bauman J, Chang JY, et al. NCCN guidelines insights: non-small cell lung cancer, version 5.2018. J Natl Compr Cancer Netw. 2018;16:807–21.
de Leng WW, Gadellaa-van Hooijdonk CG, Barendregt-Smouter FA, Koudijs MJ, Nijman I, Hinrichs JW, et al. Targeted next generation sequencing as a reliable diagnostic assay for the detection of somatic mutations in Tumours using minimal DNA amounts from formalin fixed paraffin embedded material. PLoS One. 2016;11:e0149405.
Kim EY, Cho EN, Park HS, Hong JY, Lim S, Youn JP, et al. Compound EGFR mutation is frequently detected with co-mutations of actionable genes and associated with poor clinical outcome in lung adenocarcinoma. Cancer Biol Ther. 2016;17:237–45.
Barnet MB, O'Toole S, Horvath LG, Selinger C, Yu B, Ng CC, et al. EGFR-co-mutated advanced NSCLC and response to EGFR tyrosine kinase inhibitors. J Thorac Oncol. 2017;12:585–90.
Kojima K, Konopleva M, Samudio IJ, Ruvolo V, Andreeff M. Mitogen-activated protein kinase kinase inhibition enhances nuclear proapoptotic function of p53 in acute myelogenous leukemia cells. Cancer Res. 2007;67:3210–9.
Zwang Y, Sas-Chen A, Drier Y, Shay T, Avraham R, Lauriola M, et al. Two phases of mitogenic signaling unveil roles for p53 and EGR1 in elimination of inconsistent growth signals. Mol Cell. 2011;42:524–35.
Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nature Rev Genet. 2016;17:333–51.
Kim J, Park WY, Kim NKD, Jang SJ, Chun SM, Sung CO, et al. Good laboratory standards for clinical next-generation sequencing cancer panel tests. J Pathol Transl Med. 2017;51:191–204.
Lee A, Lee SH, Jung CK, Park G, Lee KY, Choi HJ, et al. Use of the Ion AmpliSeq Cancer hotspot panel in clinical molecular pathology laboratories for analysis of solid tumours: with emphasis on validation with relevant single molecular pathology tests and the Oncomine focus assay. Pathol Res Pract. 2018;214:713–9.
Acknowledgments
To the Histopathology Laboratory personnel for the IHC experiments.
Funding
The study was not supported by external agency funds.
Author information
Authors and Affiliations
Contributions
AM conceived the idea, supervised experiments, test analysis and manuscript writing. SV analysed data and wrote the manuscript. SKS performed NGS experiments and analysis. MP did Real-Time PCR and analysis. MS supervised experiments and analysed test results. MS performed FISH experiments. UB guided and treated the patients. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
The study was approved by ‘Institutional Review Board’ (RGCIRC/ IRB/ 277/ 2019), with a waiver from patient consenting.
Consent for publication
Not applicable.
Competing interests
The authors report no conflict of interest in this work.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Venn diagram depicting the 54 genes covered by the respective panel. The genes in which alterations were detected by NGS sequencing are indicated in red. (N = 100, lung cancer cases).
Additional file 2.
Pattern and distribution of the TP53 mutations in the cohort according to (A) Mutation type (B) Mutation effect on TP53 protein sequence (C) Exon/ Intron distribution (D) Functional effect (E) SIFT prediction (IARC TP53 Database). (N = 100, lung cancer subjects).
Additional file 3.
Clinically actionable and non-actionable alterations detected by NGS in the study group.
Additional file 4.
Analysis of Progression-Free Survival (PFS), targeted therapy and mutation profile of EGFR single (S) and co-mutation (C) cases (N = 23, Lung cancer patients).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mehta, A., Vasudevan, S., Sharma, S.K. et al. Biomarker testing for advanced lung cancer by next-generation sequencing; a valid method to achieve a comprehensive glimpse at mutational landscape. Appl Cancer Res 40, 4 (2020). https://doi.org/10.1186/s41241-020-00089-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41241-020-00089-8