
Abstract
Recent findings coming from human proteome research employing mass-spectrometry and ribosomal profiling methods have provided evidence for the translation of non-annotated coding sequence (CDSs) into alternative proteins (APs). The presence of APs in many human tissues and cell lines may become an important issue in genome sciences, especially in cancer genomics where the frequency of alternative proteins seems to be 10-fold higher than normal tissues. Finding new proteins can impact medical research by filling gaps in known molecular pathways or revealing new molecular markers and therapeutic targets. Among the cellular processes possibly involved in protein diversity, alternative splicing (AS) is the most cited, and it consists of an often-regulated mechanism that generates different mRNAs from the same gene, contributing to the functional diversity of mammalian cells. In the past, evidence for AS from multi-exon genes have come mainly from expression sequence tag (EST) data; only recently has mass-spectrometry (MS) been used to investigate the translation of alternative transcripts. Exploration of human MS data has detected tens to hundreds of alternative proteins in normal tissues, and thousands in cancer cell lines, suggesting that alternative proteins may have an important role in cancer.
Analysis of MS data has revealed a vastly diverse AP repertoire, with some of this diversity being exclusively detected in cancer cells. Proteomic characterization of 20 breast cancer cell lines revealed a surprising 1,860 protein variants resulting from AS. Among these, 4 AP are clearly involved in cancer. A truncated variant of the NF- kB p65 subunit, a truncated form of the focal adhesion kinase PTK2 and two CD47 transmembrane receptor protein variants. Until now, little is known about the functional differences between these variants. Another cellular mechanism that possibly creates protein diversity is the alternative usage of translation initiation site (TIS). Detection of TIS is made possible by the Ribosome Profiling (RP) method. The principle of this technique is to capture mRNA translation by freezing the actively translating ribosomes onto transcripts, and then separating them by ultracentrifugation. Recently, RP was applied to mouse embryonic fibroblast cells and human HEK293 cells. The results revealed that the majority of mRNAs contain more than one translation initiation site (TIS), with more than 50% of the detected TISs mapping to alternative ORFs. In this review, we present a list of human alternative proteins validated by small and large-scale experimental methods. We also highlight that APs are probably not a secondary product of inaccurate splicing or translational process and most likely play an important role in the tumorigenic process. Thus, APs constitutes a promising research line for basic and clinical aspects of cancer.
Similar content being viewed by others
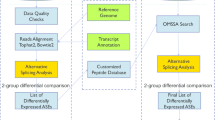

Background
Deciphering the human proteome represents an important challenge in the post-genomic era. Improvements in the current set of reference proteins can impact several aspects of health and disease studies for example by filling gaps in known molecular pathways or by revealing new molecular markers and therapeutic targets for diseases. Currently, most of the research in genomic sciences use reference protein sequences available in few databases (Ace View [1], GENCODE [2], RefSeq [3], Ensembl [4], VEGA [5], CCDS [6]). In most of cases, each gene contains one reference Open Reading Frame (ORF), commonly defined by its length (the longest one) and other criteria such the presence of domains in the predicted amino acid sequence, evolutionary conservation and number of introns [1]. In contrast, the same databases contain hundreds of thousands of distinct human gene transcripts for approximately twenty thousand genes revealing a huge diversity in the human transcriptome (e.g., http://ensembl.org/Homo_sapiens/Info/Annotation). This difference is due to the fact that almost all human genes produce several distinct transcripts resulting from molecular mechanisms such as alternative splicing [7], alternative polyadenylation [8], and alternative transcription initiation [9]. Moreover, a single transcript can have multiple ORFs (altORFs), which may originate proteins totally distinct in their amino acid composition and cellular functions (for a review see [10]). Thus, in comparison with the transcriptome, the known proteome diversity is still scarce and the assumption of a single reference protein for each gene is currently under challenge. Interestingly, among the increasing number of experimentally detected cases of alternative proteins [11,12,13], many were discovered in cancer tissues or cell lines [14] highlighting their relevance for the cancer research.
Evidences for translation of human alternatively spliced transcript
Alternative splicing (AS) is the mechanism by which the primary transcript (primary RNA) of a gene is processed in different ways resulting into distinct mature transcripts or messenger RNAs (mRNAs) [7, 15, 16]. Currently we know that practically all multi-exon human genes produce at least two differently spliced mRNA transcripts [17]. The most abundant type of AS event in mammals is exon-skipping [7, 17], a process that results in the complete presence or absence of an exon in mature transcripts (Fig. 1). Despite the widespread occurrence of AS in different physiological or pathogenic conditions [18,19,20,21], the functional relevance of such mechanisms is still under debate [22, 23]. One of the most controversial issues concerns the fraction of the non-reference transcriptome that is actually translated into proteins. Since its discovery in the 70's, alternative splicing was interpreted as a cellular mechanism that could increase the diversity of proteins [24]. However, only recently has this prediction been tested, mainly through mass-spectrometry (MS) experiments. The task of finding AP is complicated by some limitations of MS method such as the detection of proteins expressed in low levels. Moreover, a mass spectrum can be viewed as a “signature” of mass values that allows the identification a peptide, which is just a fragment of the whole protein. Additionally, the mass spectra are usually compared against peptides from a database of reference proteins. These proteins are inferred by several criteria including, in many cases, only proteins longer than 150 amino acids [1]. Thus, the chosen protein sequence database is central to the success of MS-based protein identification. All these aspects should be accounted when interpreting the relative low number of experimental validated alternative proteins so far.

Diagram showing the computational strategy applied to search an exon-skipping alternative splicing event in mass spectrometry data
Exploration of human mass spectrometry data has detected tens to hundreds of alternative proteins in normal tissues and thousands in cancer cell lines. One of the first attempts to investigate the human proteome was described in [25]. These authors collected human ESTs data and proposed a computational method to reconstruct the mRNAs. The mRNAs were then translated and searched against mass-spectrometry data. In total, these authors detected 20 instances of alternative proteins. In another study, Ezkurdia et al. [11] used public mass-spectrometry data to search against protein reference sequences from public databases. These authors detected splicing isoforms for 150 human genes, which in most of cases, differed only slightly from the reference proteins. However, for three genes (CUX1, NEBL, and MACF1), the APs differed in a considerable part compared to the full reference sequence. Moreover, they found that these protein regions, modified by alternative splicing, contain functional domains, suggesting that important cellular functions may be affected.
In 2013, Sheynkman et al. [12] performed RNAseq and mass-spectrometry analyses on the same cell population (Jurkat cells). By using a customized protein database, these authors discovered 57 peptides that align to exon-exon junctions created by alternative splicing events (Fig. 1). Of these, 12 were exon-skipping events. In a later study, Ramalho et al. [13] also searched public mass-spectrometry data for evidence of alternative proteins. In this work, hundreds of exon-skipping events, previously found by RNA-Seq, were used in the creation of a customized mRNA sequence database. These mRNAs were then translated into amino acid sequences and searched against millions of mass-spectra from public repositories. These authors detected signatures of exon-skipping events in proteins sequences from 14 human genes. Interestingly, the majority of theses exon-skipping events was present in different vertebrates species suggesting that they are not just splicing errors but may play a role in some cellular function. Moreover, among the 14 detected cases, four resulted in protein sequences that were much shorter than the reference sequences (truncated forms) and differ in amino acid composition at a terminal end.
Several of the exon-skipping events detected at the protein level by Ramalho et al. 2015 had been previously detected at the transcript level by distinct methods and authors and eight events (in the following genes: IMMT [26], COL6A3 [20], FN1 [21], TSC2 [27], CLIP170 [28], THYN1 [29], Junctin [30], Ktn1 [31]) had been previously detected not only by the recent high-throughput RNA sequencing but also by exon-array, quantitative real-time RT-PCR, and Southern and Northern Blot assays. Some of theses exon-skipping events were described as tissue-specific and/or associated with tumor tissues.
Alternative proteins in breast cancer
Recently, Lawrence et al., 2015 [14] used mass-spectrometry to conduct a proteomic characterization of 20 breast cancer cell lines and 4 TNBC tumor samples in order to characterize the proteomes and identify molecular diagnostic markers to improve drug selection. Among these cell lines, 16 were from the triple-negative breast cancer (TNBC) subtype characterized by the absence of three cellular receptors: estrogen receptor (ESR1), progesterone receptor (PGR), and human epidermal growth factor receptor-2 (ERBB2). TNBC is an object of great interest in the breast cancer field because it tends to be a more aggressive tumor subtype, correlated with worse prognosis than hormonal receptor-positive subtypes as well as is disproportionally diagnosed in women with pathogenic mutation in BRCA1 gene [32, 33]. Although a Poly (ADP-ribose) polymerase (PARP) inhibitor, Olaparib, has been approved for the treatment of advanced ovarian cancer with BRCA1 or BRCA2 mutation, only subsets of TNBC show sensitivity to this target therapy [34, 35] and, consequently, its efficiency in TNBC treatment remains uncertain.
Lawrence et al., identified 12,775 distinct proteins encoded by 11,466 genes (protein false discovery rate [FDR] < 1%). By using hierarchical clustering of protein expression measures, the cell lines were grouped in four clusters corresponding to the major molecular subtypes previously defined by mRNA expression arrays and morphological studies.
Gene ontology analysis from each cluster revealed that luminal-like cells expressed higher levels of proteins associated with proliferation, such as cell cycle, growth factor signaling, metabolism, and DNA damage repair mechanisms. TNBC cell types, particularly the tumors samples and more invasive cell lines, showed an overexpression of proteins associated with metastasis, such as ECM-receptor interaction, cell adhesion, and angiogenesis. Besides EGFR, ERBB2, ESR1 and PGR, which are already routine clinical targets in breast cancer, they found ephrin type A receptors to be highly overexpressed in many TNBC cell lines compared to luminal-like cells.
Surprisingly, 1,860 protein variants resulting from alternative splicing were found in the proteome of these cells. Furthermore, this relative high number of alternative proteins seems to be an underestimate because only isoforms already present in the reference database UniProt were considered in the search against mass spectra. Regarding proteins involved in cancer, they found a truncated variant (with a premature stop codon) for the p65 subunit of the NF-kB transcription factor. This variant lacks regulatory regions that directly affect its transcriptional activity and was detected in two cell lines and in all tumor samples as highly expressed. Additionally, two alternative splicing variants of the CD47 protein were detected in two cell lines (DU4475 and MCF7). CD47 is a G protein-coupled receptor with five membrane-spanning domains that participates in the integrin signaling and is a tumor antigen. The two protein variants differ in the cytoplasmic tail, probably resulting in distinct intracellular signaling. Until now, little is known about functional differences between these variants. Lastly, a truncated form of the focal adhesion kinase PTK2 (as well as the reference form) was detected in most cell lines analyzed. The truncate form lacks the FERM (4.1-Ezrin-Radixin-Moesin) domain that regulates PTK2 localization and interaction with other proteins.
Ribosomal profiling brings further evidence for non-canonical proteins
There is a much larger body of genome-wide evidence for the translation of non-canonical transcripts when results from the ribosome profiling method are considered [36,37,38]. This technique is used to quantify the translation state of specific mRNAs, and the idea behind it is to capture mRNA molecules in the translation process by freezing actively translating ribosomes on different transcripts, and then separating the resulting polyribosomes by ultracentrifugation on a sucrose gradient. This process allows for the identification of highly translated (bound by several ribosomes), poorly translated (bound by one or two ribosomes) and non-translated transcripts [39].
Ribosomal profiling applied to mouse embryonic fibroblast cells and human HEK293 cells revealed that the majority of mRNAs contain more than one translation initiation site (TIS) and >50% of detected TISs map to alternative ORFs [36, 37].
Recently, a machine learning approach (RibORF) was used in the interpretation of ribosomal profiling data collected from fibroblast and breast epithelial cell lines [38]. This study showed that ~40% of so-called long non-coding RNA (lncRNAs) and pseudogene RNAs are translated in vivo, and hence are not truly non-coding RNAs. Interestingly, the translated lncRNA and pseudogene peptides have median lengths of 69 and 92 amino acids, respectively, which are shorter than most of the proteins sequences available in the main reference databases.
Differently from canonical mRNAs, where the longest candidate ORFs are virtually always translated, lncRNAs have their longest ORFs translated in only 56% of cases. Moreover, most lncRNA peptides (92%) do not contain protein domains annotated on Pfam database.
Conversely, among transcribed pseudogenes (~3% of all annotated human pseudogene), 19% are translated into peptides longer than 100 amino acids and of these, 80% contain at least one protein domain.
Alternative proteins as therapeutic targets
The relevance of alternative splicing in cancer has been extensively discussed in the literature and different aspects has been highlighted, for example, the differential expression of alternatively spliced transcripts in cancer or the impact of somatic mutations in splice sites, in splicing regulatory motifs or in the core and auxiliary factors of the spliceosome (for recent reviews see [16, 40]). Since 1996, certain bacterial compounds (FR901464, herboxidienes and pladienolides)—extracted from genus Pseudomonas and Streptomyces—are known as cytotoxins which arrest cell cycle in the G1 and G2/M phases [41,42,43,44]. Despite the promising anticancer properties of these compounds they were chemically unstable and thus unsuitable for therapy. In 2007, some analogs of these compounds, most notably E7107 [45] (an analog of pladienolide B), spliceostatin A [46] (SSA; an analog of FR901464) and the sudemycins [47] were developed with improved stability.
Further studies demonstrated that the SSA and E7107 impair pre-mRNA splicing in a dose- and time-dependent manner through binding to the Splicing factor 3b (SF3B), a protein complex that is a component of U2 snRNP. U2 is an essential splicing factor, directly involved with splice site recognition [45, 46]. Importantly, these works identified that although most unspliced pre-mRNA are retained into the cell nucleus, a minor fraction goes to cytoplasm and thus are able to be translated into new proteins. Moreover, the treatment with these compounds resulted in the production of a truncated but functional form of the cell cycle inhibitor p27 (encoded by CDKN1B) that is more stable than normal because it lacks a C-terminal domain necessary for its normal degradation.
Further evidence for the central role of the SF3B splicing factor in the cell cycle arrest came from cell lines of colorectal cancer that acquired resistance to Pladienolide B. By using RNA-seq in both resistant and parental cells it was found that resistant cells acquired a point mutation in SF3B1 (Splicing factor 3b subunit 1) (SF3B1R1074H) that reduce its binding affinity to the compounds [48].
Another example of drug resistance induced by alternative proteins came from human melanomas. Vemurafenib is a potent RAF kinase inhibitor with remarkable clinical activity in some cases of melanoma tumors that harbor a common BRAF mutation (V600E), which constitutively activates downstream MAPK signaling. However, a splice variant of BRAF that lacks the RAS-binding domain (RBD) confers resistance to this drug [49]. Additionally, it was observed in a human melanoma cell line that an intronic mutation in BRAF leads to resistance to the BRAF inhibitor Vemurafenib. This mutation is associated with an in-frame skipping of exons 3–5, which encodes the RBD. Remarkably, the use of SSA restores the inclusion of these exons and can revert the Vemurafenib resistance both in vitro and in vivo [50].
Conclusions
The assumption of a single coding sequence (CDS) per gene is under challenge by increasing evidences that, in mammals, two or more proteins can be translated from the same mRNA and that the resulting proteins can interacts affecting the known gene function. Additionally, recent findings in human proteome research, mainly through the use of mass-spectrometry and ribosomal profiling methods, have brought evidence for the translation of non-annotated CDSs, some of them produced by alternative splicing events. The growing evidence for the presence of APs in distinct human tumor tissues and cell lines points to an important issue for genomic sciences and oncology that should be considered in the near future. Research on breast tumor tissues and derived cell lines have shown, for example, that these samples exhibit an at least 10-fold higher frequency of alternative proteins than normal counterparts. The relevance of alternative proteins as therapeutic targets in cancer is exemplified by the alternative p27 protein, a cell cycle inhibitor with more stable function than its normal counterpart, as well as by an alternative B-raf protein that lacks the RAS-binding domain (RBD) and confers resistance to Vemurafenib in melanoma. These findings suggest that the alternative proteome in cancer is a promising research field with the potential to reveal new cancer biomarkers, molecular pathways and therapeutic targets. Moreover, RNA genes, the non-coding ones, may be re-annotated if there is evidence that some of them are in fact translated into new proteins.
Remarkably, the translation of alternative ORFs, alternatively spliced transcripts and long non-coding RNA per se do not indicate functional relevance, and determining their function in different physiological and pathological conditions is fundamental. Thus, a central question that should be addressed in this new field of investigation is if the alternative proteins are sufficiently stable, if they can be detected by other methods and, finally, if they play a cellular role. The investigation of alternative proteins may reveal new perspectives in cancer research, and evolutionary conserved altORFs and alternatively splicing transcripts might be great candidates to start with.
Abbreviations
- altORFs:
-
ORFs not present in the reference databases
- AP:
-
Alternative protein. Protein not present in the reference databases, including proteins translated from alternatively spliced transcripts
- AS:
-
Alternative splicing. Non-canonical way in which a cell remove introns from a primary RNA
- CDS:
-
Coding sequence. Region of a gene sequence containing the aminoacids codons
- EST:
-
Expression sequence tag. Short sub-sequence of a cDNA sequence. Commonly used as a proxy for gene expression quantification
- FDR:
-
False discovery rate
- lncRNAs:
-
Long non-coding RNA. Non protein-coding transcripts longer than 200 nucleotides
- mRNA:
-
Messenger RNA. Protein-coding RNA transcript without introns and ready to be translated
- MS:
-
Mass spectrometry. Method of protein identification based on the mass of peptides
- ORF:
-
Open reading frame. Continuous stretch of codons that do not contains stop codon. An ORF has the potential to code for a protein or peptide
- RP:
-
Ribosome Profiling. Method that distinguishes mRNA molecules according to the number of ribosomes attached to them. Used as a proxy for translation level
- Spliceosome:
-
The molecular (RNA-protein) complex that performs the RNA splicing
- TIS:
-
Translation initiation site. First codon of a messenger RNA transcript translated by a ribosome
- TNBC:
-
Triple-negative breast cancer. Subtype of breast cancer characterized by the absence of three cellular receptors: estrogen receptor, progesterone and human epidermal growth factor
References
Thierry-Mieg D, Thierry-Mieg J. “AceView: a comprehensive cDNA-supported gene and transcripts annotation”. Genome Biol. 2006;7 Suppl 1:S12. 1–14.
Harrow J, et al. “GENCODE: the reference human genome annotation for The ENCODE Project”. Genome Res. 2012;22(9):1760–74.
Pruitt KD, et al. “RefSeq: an update on mammalian reference sequences”. Nucleic Acids Res. 2014;42(Database issue):D756–63.
Cunningham F, et al. “Ensembl 2015”. Nucleic Acids Res. 2015;43(Database issue):D662–9.
Harrow JL, et al. “The vertebrate genome annotation browser 10 years on”. Nucleic Acids Res. 2014;42(Database issue):D771–9.
Farrell CM, et al. “Current status and new features of the consensus coding sequence database”. Nucleic Acids Res. 2014;42(Database issue):D865–72.
Kim E, Goren A, Ast G. “Alternative splicing: current perspectives”. Bioessays. 2008;30(1):38–47.
Elkon R, Ugalde AP, Agami R. “Alternative cleavage and polyadenylation: extent, regulation and function”. Nat Rev Genet. 2013;14(7):496–506.
Kapranov P, Willingham AT, Gingeras TR. “Genome-wide transcription and the implications for genomic organization”. Nat Rev Genet. 2007;8(6):413–23.
Mouilleron H, Delcourt V, Roucou X. “Death of a dogma: eukaryotic mRNAs can code for more than one protein”. Nucleic Acids Res. 2016;44(1):14–23.
Ezkurdia I, et al. “Comparative proteomics reveals a significant bias toward alternative protein isoforms with conserved structure and function”. Mol Biol Evol. 2012;29(9):2265–83.
Sheynkman GM, et al. “Discovery and mass spectrometric analysis of novel splice-junction peptides using RNA-Seq”. Mol Cell Proteomics. 2013;12(8):2341–53.
Ramalho, R., et al., “Proteomic evidence for in-frame and out-of-frame alternatively spliced isoforms in human and mouse”. IEEE/ACM Trans Comput Biol Bioinform, 2015. [Epub ahead of print].
Lawrence RT, et al. “The proteomic landscape of triple-negative breast cancer”. Cell Rep. 2015;11(4):630–44.
House AE, Lynch KW. “Regulation of alternative splicing: more than just the ABCs”. J Biol Chem. 2008;283(3):1217–21.
Dvinge H, et al. “RNA splicing factors as oncoproteins and tumour suppressors”. Nat Rev Cancer. 2016;16(7):413–30.
Wang ET, et al. “Alternative isoform regulation in human tissue transcriptomes”. Nature. 2008;456(7221):470–6.
Carraro DM, et al. “Poly (A)+ transcriptome assessment of ERBB2-induced alterations in breast cell lines”. Plos One. 2011;6(6):e21022.
Ferreira EN, et al. “Alternative splicing enriched cDNA libraries identify breast cancer-associated transcripts”. BMC Genomics. 2010;11 Suppl 5:S4.
Thorsen K, et al. Alternative splicing in colon, bladder, and prostate cancer identified by exon array analysis”. Mol Cell Proteomics. 2008;7(7):1214–24.
Gardina PJ, et al. “Alternative splicing and differential gene expression in colon cancer detected by a whole genome exon array”. BMC Genomics. 2006;7:325.
Kelemen O, et al. “Function of alternative splicing”. Gene. 2013;514(1):1–30.
Pickrell JK, et al. “Noisy splicing drives mRNA isoform diversity in human cells”. Plos Genet. 2010;6(12):e1001236.
Gilbert W. “Why genes in pieces?”. Nature. 1978;271(5645):501.
Tanner S, et al. “Improving gene annotation using peptide mass spectrometry”. Genome Res. 2007;17(2):231–9.
Shah SH, Pallas JA. “Identifying differential exon splicing using linear models and correlation coefficients”. BMC Bioinformatics. 2009;10:26.
Kobayashi T, et al. “cDNA structure, alternative splicing and exon-intron organization of the predisposing tuberous sclerosis (Tsc2) gene of the Eker rat model”. Nucleic Acids Res. 1995;23(14):2608–13.
Griparic L, Volosky JM, Keller 3rd TC. “Cloning and expression of chicken CLIP-170 and restin isoforms”. Gene. 1998;206(2):195–208.
Miyaji H, et al. “Molecular cloning and characterization of the mouse thymocyte protein gene”. Gene. 2002;297(1–2):189–96.
Hong CS, et al. “Molecular cloning and characterization of mouse cardiac junctate isoforms”. Biochem Biophys Res Commun. 2001;289(4):882–7.
Leung E, et al. “Cloning of novel kinectin splice variants with alternative C-termini: structure, distribution and evolution of mouse kinectin”. Immunol Cell Biol. 1996;74(5):421–33.
Carraro DM, et al. “Comprehensive analysis of BRCA1, BRCA2 and TP53 germline mutation and tumor characterization: a portrait of early-onset breast cancer in Brazil”. Plos One. 2013;8(3):e57581.
Fackenthal JD, Olopade OI. “Breast cancer risk associated with BRCA1 and BRCA2 in diverse populations”. Nat Rev Cancer. 2007;7(12):937–48.
Fong PC, et al. “Inhibition of poly (ADP-ribose) polymerase in tumors from BRCA mutation carriers”. N Engl J Med. 2009;361(2):123–34.
Silver DP, et al. “Efficacy of neoadjuvant Cisplatin in triple-negative breast cancer”. J Clin Oncol. 2010;28(7):1145–53.
Ingolia NT, et al. “Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes”. Cell Rep. 2014;8(5):1365–79.
Ingolia NT, Lareau LF, Weissman JS. “Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes”. Cell. 2011;147(4):789–802.
Ji Z, et al. “Many lncRNAs, 5'UTRs, and pseudogenes are translated and some are likely to express functional proteins”. Elife. 2015;4:e08890.
Faye MD, Graber TE, Holcik M. “Assessment of selective mRNA translation in mammalian cells by polysome profiling”. J Vis Exp. 2014;92:e52295.
Sebestyen E, et al. “Large-scale analysis of genome and transcriptome alterations in multiple tumors unveils novel cancer-relevant splicing networks”. Genome Res. 2016;26(6):732–44.
Nakajima H, et al. “New antitumor substances, FR901463, FR901464 and FR901465. I. Taxonomy, fermentation, isolation, physico-chemical properties and biological activities”. J Antibiot (Tokyo). 1996;49(12):1196–203.
Nakajima H, et al. “New antitumor substances, FR901463, FR901464 and FR901465. II. Activities against experimental tumors in mice and mechanism of action”. J Antibiot (Tokyo). 1996;49(12):1204–11.
Mizui Y, et al. “Pladienolides, new substances from culture of Streptomyces platensis Mer-11107. III. In vitro and in vivo antitumor activities”. J Antibiot (Tokyo). 2004;57(3):188–96.
Sakai T, et al. “Pladienolides, new substances from culture of Streptomyces platensis Mer-11107. II. Physico-chemical properties and structure elucidation”. J Antibiot (Tokyo). 2004;57(3):180–7.
Kotake Y, et al. “Splicing factor SF3b as a target of the antitumor natural product pladienolide”. Nat Chem Biol. 2007;3(9):570–5.
Kaida D, et al. “Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA”. Nat Chem Biol. 2007;3(9):576–83.
Fan L, et al. “Sudemycins, novel small molecule analogues of FR901464, induce alternative gene splicing”. ACS Chem Biol. 2011;6(6):582–9.
Yokoi A, et al. “Biological validation that SF3b is a target of the antitumor macrolide pladienolide”. FEBS J. 2011;278(24):4870–80.
Poulikakos PI, et al. “RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF (V600E)”. Nature. 2011;480(7377):387–90.
Salton M, et al. “Inhibition of vemurafenib-resistant melanoma by interference with pre-mRNA splicing”. Nat Commun. 2015;6:7103.
Acknowledgements
We would like to thank both Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) for the grant 2013/23277-8 to Dirce Carraro and Fábio Kuriki Mendes for kindly reading this article and providing helpful comments.
Funding
Not applicable.
Availability of data and materials
Not applicable.
Authors’ contributions
RFR collects and review all the bibliography. DMC and RFR equally contributed to sections design and manuscript writing. Both authors read and approved the final manuscript.
Competing interests
The author declares that have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ramalho, R.F., Carraro, D.M. Increasing evidence for the presence of alternative proteins in human tissues and cell lines. Appl Cancer Res 37, 10 (2017). https://doi.org/10.1186/s41241-017-0013-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41241-017-0013-0