
Abstract
Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) is the causative virus of pandemic acute respiratory disease called coronavirus disease 2019 (COVID-19). Most of the infected individuals have asymptomatic or mild symptoms, but some patients show severe and critical systemic inflammation including tissue damage and multi-organ failures. Immune responses to the pathogen determine clinical course. In general, the activation of innate immune responses is mediated by host pattern-recognition receptors (PRRs) that recognize pathogen-associated molecular patterns (PAMPs) as well as host damage-associated molecular patterns (DAMPs), which results in the activation of the downstream gene induction programs of types I and III interferons (IFNs) and proinflammatory cytokines for inducing antiviral activity. However, the excessive activation of these responses may lead to deleterious inflammation. Here, we review the recent advances in our understanding of innate immune responses to SARS-CoV-2 infection, particularly in terms of innate recognition and the subsequent inflammation underlying COVID-19 immunopathology.
Similar content being viewed by others

Background
Coronaviruses, named for the crown-like spikes on their surface, are common pathogens of humans and animals. In the 1960s, the first coronaviruses that infect humans were identified [1]. Four types of human coronaviruses (human coronavirus (HCoV)-229E, HCoV-NL63, HCoV-OC43, and HCoV-HKU1) are prevalent and typically cause some 10 to 15% of common cold symptoms in immunocompetent individuals [2]. SARS-CoV and Middle East respiratory syndrome coronavirus (MERS-CoV), which have confirmed in human in 2002 and 2012, respectively, are highly pathogenic provoking regional and global outbreaks [3,4,5]. Since December 2019, SARS-CoV-2 has emerged as a major public health crisis, resulting in devastating deaths worldwide [6]. COVID-19 is characterized by both upper and lower respiratory tract infections, which leads to either asymptomatic or a variety of clinical symptoms including cough, fever, and pneumonia, along with other complications like diarrhea and multi-organ failure [7,8,9,10,11,12,13]. Risk of severe disease among COVID-19 patients is highly influenced by patients with older age, smoking, chronic obstructive pulmonary disease (COPD), cardiovascular disease, diabetes, obese, cancer, hypertension, and acute kidney injury [14,15,16,17].
Recognition of invading viruses by PRRs is the first step of host defense against infection to activate innate immune signaling cascades, which result in the production of types I and III IFNs, proinflammatory cytokines, and chemokines. Types I and III IFNs are critical cytokines to confer cells with anti-viral state [18]. Genetical disruption of these IFNs and the related genes shows higher risk of severe COVID-19 in humans [19,20,21,22]. Additionally, a limited and delayed IFN responses in human patients result in exacerbated expression of proinflammatory cytokine, which contribute to severe SARS-CoV-2 symptomatology [23,24,25,26]. Indeed, studies of patients with COVID-19 have reported the increment of inflammatory monocytes and neutrophils, a sharp decrease in lymphocytes, and an inflammatory milieu containing interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF) in severe disease [7, 24, 27,28,29,30,31].
In this review, we review recent findings on the molecular mechanisms of innate immune activation, in particular, by focusing on innate recognition of SARS-CoV-2.
The life cycle of SARS-CoV-2 in host cells
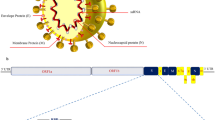
SARS-CoV-2 is an enveloped virus containing about 30,000 nucleotides of single-stranded, positive-sense genomic RNA [(+)gRNA] complexed with nucleocapsid (N) protein and expresses the viral proteins, spike (S), envelope (E), and membrane (M) structural proteins on its envelope [32, 33]. SARS-CoV-2 primarily infects the nasal and respiratory tract such as nasal epithelial cells, bronchial epithelial cells, alveolar epithelial type II cells, and vascular endothelial cells [34,35,36,37,38]. The S glycoprotein mediates the viral entry into the host cells through its binding of the receptor angiotensin-converting enzyme 2 (ACE2), followed by proteolytic cleavage by host proteases such as transmembrane protease serine 2 (TMPRSS2) at the cell surface or by cathepsin L in the endosome [39,40,41]. Other host proteins such as neuropilin-1 (NRP1), C-type lectins, furin, KIM-1, and AXL were also identified as cellular cofactors for viral entry [42,43,44,45,46]. In addition, Fcγ receptors and CD147 were reported as the other receptors for SARS-CoV-2 infection in monocytes/macrophages and T cells, respectively [31, 47].
Upon viral entry, viral membranes fuse with host membranes to introduce viral (+)gRNA into the cytoplasm [39]. By using the host machineries, the (+)gRNA is translated into large polyproteins (pp1a and pp1ab), which are then cleaved into sixteen types of nonstructural proteins (NSPs) by viral 3C-like protease Mpro [48]. Among these NSPs, NSP12 harboring RNA-dependent RNA polymerase (RdRp) catalytic activity and two accessory proteins (NSP7 and NSP8) form a complex [49] and bind to the 3′-untranslated region (3′UTR) of (+)gRNA, which initiate the continuous and discontinuous synthesis of negative-sense RNAs [(−)RNAs] for viral replication and gene expression, respectively [2, 32, 33, 50]. These (−)RNA intermediates serve as a template for the synthesis of (+)gRNA and subgenomic RNAs, and various viral structural proteins and accessory proteins are also translated. Finally, the (+)gRNA is packaged by the structural proteins to assemble progeny virions and bud to release viral particles [6, 33].
In processes of viral entry and replication, virus-derived components such as nucleic acids and the virion proteins are recognized by PRRs on cell surface or in the cytoplasm, leading to the activation of innate responses.
Sensing of SARS-CoV-2 by PRRs to activate innate immune signaling
The innate immune responses are initiated with the detection of PAMPs or DAMPs by PRRs. Based on the protein domain homology, PRRs can be classified into one of six groups consisted of Toll-like receptors (TLRs), retinoic acid-inducible gene-I (RIG-I)-like receptors (RLRs), C-type lectin receptors (CLRs), nucleotide-binding and oligomerization domain (NOD)-like receptors (NLRs), the absence in melanoma 2 (AIM2)-like receptors (ALRs), and the other types including intracellular DNA sensor, cyclic guanosine monophosphate-adenosine monophosphate (cGAMP), and synthase (cGAS) [18, 51,52,53,54]. In this section, we summarize recent reports regarding innate recognition of SARS-CoV-2 and PRR-mediated innate immune signalings during SARS-CoV-2 infection (Table 1).
TLR-mediated sensing of SARS-CoV-2
TLRs play a crucial role in the activation of innate immune responses against infection with a variety of pathogens [80]. The subcellular localization of TLRs is exclusively in endolysosome or on plasma membrane and generally transduce downstream signalings via two key adaptor molecules, myeloid differentiation factor 88 (MyD88), and Toll/IL-1 receptor domain-containing adaptor inducing IFN-β (TRIF; also known as TICAM-1) [54]. Most of TLRs, except for TLR3, use MyD88 to activate transcription factors, nuclear factor (NF)-κB, and activator protein-1 (AP-1). TLR3 and TLR4 have another adaptor protein TRIF independently of MyD88 to activate NF-κB, AP-1, and IFN regulatory factors (IRFs). TLRs are expressed preferentially in immune cells including monocytes, macrophages, neutrophils, mast cells, basophils, and dendritic cells [81, 82]. In the case of viral infection, numerous studies have shown that in the most case, viral PAMPs for TLRs include viral nucleic acids and proteins. TLR2 is involved in the recognition of viral structural proteins such as EBV-encoded dUTPase, HSV-1-encoded glycoprotein B, and hepatitis B virus capsid [83,84,85]. TLR4 senses the fusion protein of respiratory syncytial virus (RSV), glycoprotein of Ebola virus, glycoprotein G of vesicular stomatitis virus, and nonstructural protein 1 of dengue virus [86,87,88,89]. As for RNA-sensing TLRs, TLR3 has a protective role against SARS-CoV infection in mice [90]. TLR7 is required for type I IFN response during infection with mouse hepatitis virus (MHV), a murine coronavirus, and MERS-CoV in dendritic cells [91, 92].
As for the involvement of TLRs in COVID-19, a single cell-based computational method with the dataset of bronchoalveolar lavage from patients with mild and severe COVID-19 identified TLR2 as a pathogenic factor for the hyperinflammatory response [55]. In this context, it was reported that TLR2 is required for the production of inflammatory cytokines in peripheral blood mononuclear cells (PBMC) during SARS-CoV-2 infection [56]. The extracellular treatment with recombinant SARS-CoV-2 E protein but not S protein induced the proinflammatory cytokine response in bone marrow-derived macrophages (BMDM), and the E protein-triggered response was reduced in TLR2-deficient BMDM [56]. In contrast, there is a report by another research group showing that such an E protein-mediated inflammatory response was not observed in macrophages and lung epithelial cells [57]. In addition, SARS-CoV-2 S protein was shown to be rather immunostimulatory to produce proinflammatory cytokines via TLR2 and TLR4 in macrophages [57, 59, 60, 93]. In silico data indicated that TLR4, TLR6, and TLR1 possess a strong binding affinity to spike protein [61]. Genetic variations in genes encoding TLR3 and TLR7 were shown to be related to the severity of COVID-19 [19,20,21], while there is also a report showing no significant association [78]. Treatment with TLR3 and TLR7 inhibitors or siRNAs decreased the induction of type I and type III IFNs and proinflammatory cytokines after SARS-CoV-2 infection in Calu-3/MRC-5 multicellular spheroids [58]. Further studies are needed to clarify what PAMPs are directly sensed by TLRs during SARS-CoV-2 infection.
RLR-mediated sensing of SARS-CoV-2
RLRs such as RIG-I and melanoma differentiation-associated gene 5 (MDA5) are localized in the cytoplasm and recognizes ssRNA and dsRNA, which are virus-derived genomes and replication intermediates and are involved in the recognition of various types of RNA viruses. RIG-I senses RNAs carrying a 5′-triphosphate modification (3pRNA) or short-type dsRNAs in cells infected with a variety of RNA viruses such as influenza A virus, measles virus, and hepatitis C virus, which are widely known to be pathogenic to humans [94,95,96,97]. MDA5 mainly recognizes double-stranded RNAs of 3 kb or longer [97] and is required for innate immune responses against certain types of viruses such as Picornaviruses and Flaviviruses [98]. RIG-I and MDA5 consist of two caspase-recruitment domains (CARDs), DExD/H-box helicase domain (HD) and C-terminal domain (CTD). Upon RNA ligand binding via their CTD, the CARDs interact with the adaptor molecule mitochondrial antiviral signaling protein (MAVS; also known as IPS-1, VISA, or Cardif), resulting in gene transcription of types I/III IFNs and inflammatory cytokines [18, 53, 54]. It is also reported that RLRs are required for the sensing of coronaviruses: Both RIG-I and MDA5 are involved in the induction of type I IFNs and proinflammatory cytokines during infection with MHV [56, 99,100,101]. RIG-I also contributes to the inflammatory cytokine production in response to MERS-CoV infection [102]. However, although it is likely that RLRs sense RNA species derived from such coronaviruses, the detailed mechanism remains poorly understood.
The basal expression levels of the RLRs are higher in upper airway epithelial cells, macrophages, and dendritic cells of children [103], which may suggest a lower risk for developing COVID-19 in children, compared in those of adults [17]. These data also suggest that RLRs may play a role in the first-line defense against SARS-CoV-2 infection. MDA5 is most likely a candidate intracellular RNA sensor for eliciting innate immune cytokine responses against SARS-CoV-2 in lung epithelial cells. Many studies showed that silencing or knockout of MDA5 but not RIG-I results in reduced types I and III IFN responses during SARS-CoV-2 infection in a lung epithelial cell line, Calu-3 cells [62,63,64,65,66,67]. However, there is a report showing that RIG-I is also involved in cytokine responses in Calu-3 cells [66]. In addition, transfection with viral RNAs extracted from TMRPSS2-expressing Vero E6 cells infected with SARS-CoV-2 resulted in the induction of innate cytokines in HEK293 cells [68]. In this regard, we confirmed that knockdown of MDA5 but not RIG-I remarkably suppressed the induction of both types I/III IFNs and IL-6 in Calu-3 cells infected with SARS-CoV-2. But we also found that RIG-I is capable to sufficiently restrain SARS-CoV-2 replication in primary human bronchial and alveolar epithelial cells without activation of the conventional RIG-I downstream signaling [67]. The protein expression levels of RIG-I in Calu-3 cells are markedly lower than those of primary human primary bronchial and alveolar epithelial cells as well as A549 cells, which we tested. This may at least partly explain the reason why SARS-CoV-2 replication is observed in Calu-3 cells but not primary human lung epithelial cells. In the first step of viral replication, RIG-I competitively inhibits the access of viral RdRp to the 3′UTR of the viral (+)gRNA through the RIG-I HD. This RIG-I HD-mediated recognition fails to activate the conventional downstream MAVS-dependent IRF/NF-κB signaling pathways, which is in accordance with lack of cytokine induction after SARS-CoV-2 infection in primary human lung epithelial cells (Fig. 1). Consistent with this observation, SARS-CoV-2 can replicate in cells harboring low levels of RIG-I expression such as COPD patient-derived cells, which might link to acute infectious exacerbation in COPD patients [15, 104,105,106]. Therefore, in the situation where (−)RNA initiates to be transcribed from the viral (+)gRNA, MDA5 in turn play a role as an innate sensor to induce types I/III IFNs and other cytokines. Furthermore, treatment with all-trans retinoic acid (ATRA), which was originally reported to upregulate RIG-I mRNA in a human promyelocytic leukemia cell [107], significantly augments RIG-I protein expression levels in COPD patient-derived bronchial epithelial cells [67]. ATRA or possible other RIG-I inducer(s) may thus be promising agents to enhance preventive and/or therapeutic potentials of COVID-19 patients. RIG-I expression levels are one of the intrinsic determinants for the defense in human lung epithelial cells during the initial process of SARS-CoV-2 infection [67]. Thus, RLRs come into play in a stepwise manner: RIG-I is the first sentinel against SARS-CoV-2 infection in human alveolar and bronchial epithelial cells, and once SARS-CoV-2 initiates to transcribe the (−)RNA, MDA5 functions a major viral sensor to induce innate cytokine responses (Fig. 1).

Schematic model of innate recognition of SARS-CoV-2 (+)gRNA. RIG-I consists of two CARDs, HD and CTD. RIG-I conventionally senses 3pRNA or short-type dsRNA via its CTD, followed by the conformational change. And then, oligomerized RIG-I binds to its adaptor protein MAVS/IPS-1, through its CARDs, which resulted in the production of types I and III IFNs and inflammatory cytokines (left). On the other hand, upon SARS-CoV-2 infection, RIG-I preferentially senses the 3′UTR of the viral (+)gRNA through its HD but not CTD. This unconventional recognition of RIG-I fails to activate the downstream MAVS/IPS-1-dependent signaling pathways. Instead, RIG-I directly exerts an antiviral activity via competitive inhibition of the recruitment of viral RdRp to viral (+)gRNA, which blocks the first step of the RdRp-dependent transcription process. CARD, caspase recruitment domain; HD, helicase domain; CTD, C-terminal domain
CLR-mediated sensing of SARS-CoV-2
CLRs, which are generally expressed in myeloid cells such as dendritic cells, monocytes, macrophages, and neutrophils, are involved in the detection of pathogen-derived mannose, fructose, and glucan carbohydrate structure [108, 109]. Recognition of viruses by CLRs modulates myeloid cell functions including gene transcription, endocytosis, and phagocytosis, thereby regulating antigen presentation, antiviral responses, and T-cell differentiation [109]. Mincle, LSECtin, and CLEC5A act as innate sensors to induce inflammation in response to MERS-CoV, Ebola virus, and dengue virus, respectively [102, 110,111,112]. On the other hand, some CLRs have been suggested as entry receptors or attachment factors for certain viruses. DC-SIGN promotes viral entry of SARS-CoV, HIV-1, HIV-2, influenza virus, dengue virus, and Ebola virus [113,114,115,116,117,118]. In addition, L-SIGN and MBL facilitate infection with SARS-CoV and Ebola virus, respectively [119, 120].
A screening assay using ectopic expression for myeloid cell-associated receptors involved in the attachment with SARS-CoV-2 S protein identified five CLRs (DC-SIGN, L-SIGN, LSECtin, ASGR1, and CLEC10A) and Tweety family member 2 (TTYH2) [69]. These five CLRs are expressed by myeloid cells from COVID-19 individuals with hyperinflammation and are engaged in the S protein-mediated robust proinflammatory cytokine response but not actively in virus entry. On the other hand, there are reports showing that the recognition of S protein by DC-SIGN and L-SIGN promotes the entry of SARS-CoV-2 to target cells [121, 122]. Therefore, certain CLRs may play some role in the process of viral entry or the S protein-mediated cytokine responses, although further investigation needs to validate their roles.
NLR-mediated sensing of SARS-CoV-2
NLRs comprise a large family of intracellular PRRs that are composed of a central NOD and C-terminal leucine-rich repeats and play an important role in the surveillance of the intracellular environment for the presence of infection, noxious substances, and metabolic perturbations [123, 124]. Among NLRs, NLR family, pyrin domain containing 3 (NLRP3) is a well-studied PRR that forms a muti-molecular protein complex, termed inflammasome, upon its activation [125,126,127]. Its expression is predominantly observed in splenic neutrophils, macrophages, monocytes, and conventional dendritic cells [128]. The activation of NLRP3 inflammasome is a two-step process, priming and activation. The priming step is to upregulate the expression of inflammasome components such as NLRP3, caspase-1, and pro-IL-1β, through the activation of NF-κB upon exposure of inflammatory stimulation such as PAMPs, DAMPs and cytokines. The activation step occurs following the recognition of an NLRP3 activators such as efflux of potassium ion generated by a large array of stimulation including extracellular ATP, microbial agonists, and uric acid crystals. The activated NLRP3 recruits adaptor protein apoptosis-associated speck-like protein containing a CARD (ASC) and caspase-1, which ultimately leads to pyroptosis or the maturation of IL-1β and IL-18 [127]. It is reported that this NLRP3 inflammasome is activated upon infection with viruses such SARS-CoV, encephalomyocarditis virus (EMCV), and influenza virus. In SARS-CoV infection, the NLRP3 inflammasome is mainly activated by viroporins that have ion channel activity [129,130,131], and that SARS-CoV viroporins enhance viral replication and virulence [132]. Viroporin 2B of EMCV also activates NLRP3 inflammasome via the induction of calcium ion flux [133]. Moreover, the activation of NLRP3 inflammasome is triggered by not only viral protein M2 ion channel but also viral RNA during influenza virus infection [134,135,136,137]. NOD1 and NOD2 represent two well-characterized PRRs of the NLR family and recognize conserved motifs of bacterial peptidoglycan, γ-D-glutamyl-meso-diaminopimelic acid (iE-DAP), and muramyl dipeptide (MDP), respectively [138]. After NOD1 and NOD2 sense their ligands, they recruit receptor-interacting serine/threonine-protein kinase 2 (RIPK2), thereby activating the downstream NF-κB and mitogen-activated protein kinase (MAPK) pathways to induce the production of proinflammatory cytokines and antimicrobial responses [138]. Although their roles as bacterial sensors are well-established, several studies uncovered a key function of NOD1 and NOD2 in sensing both RNA and DNA viruses. NOD1 activates type I IFN response in response to Cytomegalovirus and hepatitis C virus but not RSV [139,140,141], whereas NOD2-mediated responses are observed after infection with Cytomegalovirus, influenza virus, and RSV [141,142,143]. These mechanisms of how NOD1 and NOD2 rely on the type I IFN response could be partially explained by their RNA binding properties [140, 141, 144], but it remains still largely unknown.
Elevated proinflammatory cytokines such as IL-1β and IL-18 are characteristic of patients with severe COVID-19 [24]. Several studies particularly focused on the role of NLRP3 in SARS-CoV-2 infection. The concentrations of inflammasome-related markers, IL-1β, IL-18, gasdermin D, and lactate dehydrogenase, were significantly elevated in the serum and plasma of patients with COVID-19, compared with that of healthy donors [31, 70]. In addition, the formation of NLRP3-ASC puncta was detected in monocytes derived from COVID-19 patients. The immunohistochemical staining with lung autopsies of COVID-19 patients confirmed that such puncta were observed in lung tissue-resident monocytes and macrophages [31, 70]. In vitro infection assay also showed SARS-CoV-2 engaged the NLRP3 inflammasome in human monocytes [70, 71]. These results indicated that the NLRP3 inflammasome forms in monocytes and macrophages of COVID-19 patients. Viral RNAs or proteins are proposed as PAMPs responsible for the activation of NLRP3. Treatment with GU-rich single-stranded RNA of SARS-CoV-2 sequence resulted in NLRP3-dependent production of IL-1β in human primary macrophages [72], although the detail mechanism needs further investigation. Open reading frame 3a (ORF3a), a viroporin, and N protein of SARS-CoV-2 triggered IL-1β production in an NLRP3-dependent manner [73, 74]. However, there is an inconsistent report showing that the N protein inhibits the gasdermin D cleavage and IL-1β production in THP-1 cells and human primary monocytes [79]. The stimulation with recombinant SARS-CoV-2 S protein induces NLRP3 activation in macrophages derived from COVID-19 patients but not healthy individuals [75]. In this respect, the authors identified that TLR2 is also required possibly as the priming step for this NLRP3-dependent activation. In support of this, an independent study showed that NLRP3 and IL-1β mRNAs were induced by the treatment with recombinant SARS-CoV-2 E protein in a TLR2-dependent manner, in PBMC and BMDM [56]. Furthermore, TLR8 likely mediates the induction of NLRP3 expression through the detection of GU-rich SARS-CoV-2 sequence RNA [72]. Overall, the NLRP3 inflammasome is activated by various viral PAMPs, and its priming step is coordinated at least by TLRs during SARS-CoV-2 infection. IAnother NLR, NOD1, was identified as a positive regulator for IFN-β mRNA induction in Calu-3 cells during SARS-CoV-2 infection by siRNA-based screening [62], although the detail mechanism is still unclear. Considering that NOD1 modulates MDA5-MAVS complex formation [144], NOD1 may confer an enhancing effect of MDA5-mediated IFN response against SARS-CoV-2 infection.
ALR-mediated sensing of SARS-CoV-2
ALRs are intracellular innate immune sensors responsible for the detection of DNA and comprise several members of the PYHIN family including AIM2 and IFN-γ-inducible protein 16 (IFI16) [145,146,147]. AIM2 recognizes cytosolic DNA via its hematopoietic IFN-inducible nuclear protein (HIN) domain, thus inducing the recruitment of the adaptor protein ASC in monocytes and macrophages [148, 149]. This ASC recruitment allows for the formation of a large multi-protein complex, which mediates caspase-1 activation and the maturation of IL-1β and IL-18. The roles of AIM2 were well-established during infection with bacterial pathogens and DNA viruses such as herpes simplex virus-1 (HSV-1), Cytomegalovirus, and Vaccinia virus [148, 150,151,152]. On the other hand, in the case of RNA virus infection such as influenza virus, it is reported that AIM2 inflammasome activation is triggered through the accumulation of the oxidized mitochondrial DNA in the cytosol [153]. Based on these findings, both DNA and RNA viruses are likely to activate AIM2 inflammasome.
It is shown that the AIM2 inflammasome is activated in monocytes from patient with COVID-19 [31]. As mentioned above, the expression of inflammatory cytokines including IL-18 is actually upregulated in serum and plasma from severe COVID-19 patients [24, 31, 70]. Analysis with confocal microscopy confirmed ASC specks co-localized with AIM2 in COVID-19 monocytes. It is suggested that AIM2 may sense DNA released from mitochondria [154]; however, further detailed analysis is needed to understand how AIM2 is activated during SARS-CoV-2 infection. In addition, the AIM2-ASC specks also co-localizes with NLRP3, suggesting AIM2 and NLRP3 make the same inflammasome. It would be interesting to investigate the relevance of AIM2 to NLRP3 activation during SARS-CoV-2 infection.
cGAS-mediated activation of innate signaling by SARS-CoV-2 infection
cGAS is an essential cytosolic DNA sensor to trigger the innate immune responses against microbial infections. Following the binding of dsDNA, cGAS catalyzes the synthesis of a second messenger, cGAMP, in the presence of GTP and ATP, which subsequently binds to activate the adaptor molecule stimulator of interferon genes (STING) [155,156,157]. And then, STING recruits and activates TANK-binding kinase 1 (TBK1) as well as NF-κB and IRFs, to induce the production of types I/III IFNs and proinflammatory cytokines. In addition to the sensing of exogenous DNA, cGAS can also be activated by endogenous DNA, including DNA released from mitochondria and extranuclear chromatin damaged by genotoxic stress, in autoinflammatory disorders, inflammation, cellular senescence, cancer, and DNA damage response [158,159,160,161,162,163,164,165,166,167,168,169,170]. Of note, it is demonstrated that the release of mitochondrial DNA to cytosol is observed during infection with dengue virus, one of Flaviviruses, resulting the activation of cGAS [171]. In this context, cGAS has shown a striking antiviral property against not only DNA viruses such as HSV-1, Vaccinia virus, and Cytomegalovirus but also positive-strand ssRNA viruses including members of Flaviviruses [171,172,173,174]. Thus, cGAS activates the downstream signaling through the sensing of viral DNAs or the indirect effects of virus infection that causes mis-localization of self-DNA.
For SARS-CoV-2 infection, lung epithelial cells are the primary site of infection. Experiments with STING-deficient Calu-3 cells suggest that STING-mediated signaling pathway is largely dispensable for types I and III IFN response and the control of viral replication during SARS-CoV-2 infection [64, 65]. However, subsequent studies showed that knockdown of cGAS reduced the mRNA induction of TNF and IL-6 in response to SARS-CoV-2 infection in ACE2-expressing A549 cells [76]. Importantly, in this case, cGAS-STING pathway drives NF-κB-dependent proinflammatory cytokine induction but fails to produce substantial amounts of IFNs, which may partly explain the shift toward an aberrant proinflammatory response [175]. Additionally, SARS-CoV-2 S protein-induced cell fusion causes DNA damage response and induces the formation of micronuclei that are sensed by cGAS, resulting in the induction of IFN-β in ACE2-expressing A549 cells, HEK293T cells, and HeLa cells [77]. Further studies with human primary lung epithelial cells and in vivo models will be required to clarify the anti-SARS-CoV-2 role of cGAS in lung epithelial cells. Besides lung epithelial cells, Domizio D. J. et al. showed that cGAS-STING pathway-driven type I IFN signature is mediated by macrophages adjacent to the areas of endothelial cell damage [38, 175, 176]. Endothelial cells containing damaged mitochondria were detected in skin biopsies and lung tissues from patients with COVID-19. In particular, increased cGAMP levels and phosphorylated STING (STING activation marker) were observed in perivascular macrophages in the skin lesions of COVID-19 patients. Additionally, endothelial cells showed STING-dependent type I IFN response and cell death during SARS-CoV-2 infection. These observations likely explain the mechanisms underlying the aberrant immunopathology at least during the late phase of SARS-CoV-2 infection [38, 175, 176].
A stepwise model of innate sensor-mediated sensing of PAMPs and DAMPs during SARS-CoV-2
PRR-mediated recognition of invading viruses is the first step of host defense, activating antiviral and inflammatory responses. However, the excessive activation leads to deleterious systemic inflammation. While most individuals infected with SARS-CoV-2 show asymptomatic or mild symptoms, some patients experience severe disease with aberrant inflammation including tissue damage and multi-organ failures. The wide spectrum of clinical manifestation of COVID-19 patients suggests that individual immune responses to SARS-CoV-2 may critically determine the clinical course. Here, we will discuss how PAMPs and DAMPs are spatiotemporally sensed by an array of PRRs in certain cell types, particularly in terms of innate activation.
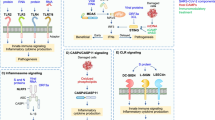
It has been proposed that SARS-CoV-2 infection primarily targets the respiratory tract [37]. In bronchial and alveolar epithelial cells infected with SARS-CoV-2, RIG-I first senses (+)gRNA of SARS-CoV-2 and acts as a direct restraining factor without the production of types I/III IFNs and proinflammatory cytokines, in an unconventional manner. This may link to asymptomatic (or mild) manifestation of COVID-19 patients. In this respect, the balance between RIG-I expression levels and the load of invading viruses would regulate the fate of viral replication (Fig. 2A), especially in the early phase of infection. If RIG-I could not recognize the (+)gRNA in some cells with downregulated RIG-I expression such as Calu-3 cells and COPD patient-derived lung epithelial cells, MDA5 in turn senses viral (−)RNA transcribed from the (+)gRNA by viral RdRp. Also, cGAS may sense host damaged DNA as a DAMP in the infected epithelial cells. Thus, MDA5 and cGAS mainly contribute to the production of types I/III IFNs and proinflammatory cytokines in epithelial cells in the early phase of infection (Fig. 2B). On the other hand, a variety of PRRs such as TLRs, CLRs, NLRs, and AIM2 come into play for the recognition of viral PAMPs as well as host DAMPs, particularly in macrophages and monocytes, which may likely lead to exacerbated proinflammatory cytokine production with extensive infiltrations of inflammatory cells in the respiratory tract in the late phase (Fig. 2C). Such multiple innate sensor-mediated inflammation disrupts mitochondrial homeostasis of vascular endothelium, resulting in the cytosolic accumulation of mitochondrial DNA, which activates cGAS-STING pathway for inducing innate cytokine responses and cell death. The dead vascular endothelial cells are engulfed by macrophages at the perivascular lesions, which also lead to cGAS activation. This innate sensor-mediated signaling circuit may contribute to sustained, dysregulated cytokine responses (Fig. 2D), which may explain COVID-19 immunopathology at least in the late phage of infection.

Stepwise model of PRR-mediated activation of innate immune response in lung tissues during SARS-CoV-2 infection. In the first step, RIG-I senses (+)gRNA of SARS-CoV-2 that is released from a viral particle and directly inhibits viral replication without activation of the conventional RIG-I downstream signaling A. In the condition of reduced RIG-I expression or in the epithelial cells with low/no levels of RIG-I expression, viral (−)RNAs initiate to be transcribed from (+)gRNA by viral RdRp. Then, MDA5 in turn plays a role as an innate sensor to induce the expression of types I/III IFNs and proinflammatory cytokines. This signaling might result in cell damage, leading to activation of cGAS pathway via possibly the sensing of host nuclear or mitochondrial DNA B. In immune cells such as macrophages/monocytes, TLRs, CLRs, NLRP3, and AIM2 function as major innate sensors to recognize viral proteins, nucleic acids, and host DAMPs, resulting in innate cytokine responses C. These innate inflammatory responses induce the death of neighboring endothelial cells, leading to the activation of cGAS pathway via its mitochondrial DNA in the cells. The dead endothelial cells are engulfed by perivascular macrophages, and then the host damaged DNA activates cGAS pathway, which induces type I IFNs and proinflammatory cytokines D. This cGAS machinery may initiate a self-perpetuating loop of the sterile inflammation, causing the detrimental inflammation in late phase of infection
In summary, we would propose that PRR-mediated innate antiviral defense during SARS-CoV-2 infection consists of at least three steps as follows: (1) RIG-I-mediated direct antiviral responses cell without cytokine response, (2) PAMP-dominant activation of innate signaling with cytokine response, and (3) DAMP-dominant activation of innate signaling with dysregulated cytokine response. While innate immune cytokines play a beneficial role for the successful clearance of invading viruses in the early stage, their excessive production in the late stage may contribute to aggravate COVID-19 immunopathology.
Conclusions
Not only nucleic acid sensors but also other types of PRRs have been reported to be involved in the activation of innate cytokine responses against SARS-CoV-2. In the early phase of SARS-CoV-2 infection, both types I and III IFNs can exert their antiviral activities. In this respect, it is also reported that types I and III IFNs are important cytokines to inhibit viral infection by inducing antiviral genes including anti-SARS-CoV-2 genes such as LY6E and BST2 [177, 178]. However, severe COVID-19 patients fail to suppress viral replication in the early phase of infection due to insufficient and delayed types I and III IFN responses, which results in exacerbated proinflammatory cytokine production in the late phase [24,25,26]. Several clinical trials showed that early administration of types I and III IFNs significantly prevented the clinical deterioration and reduced the duration of detectable virus [179,180,181,182], whereas later administration of type I IFNs was associated with increased mortality [179, 180], highlighting the opposing effects of type I IFNs for host protection and immunopathology. This harmful effect may be partially explained by a recent report showing that both types I and III IFNs disrupt lung epithelial repair during recovery from viral infection [183]. On the other hand, circulating neutralizing autoantibodies against type I IFNs are found in about 10% of patients with critical COVID-19 and in elderly individuals but not in young individuals with asymptomatic or mild SARS-CoV-2 infection [184,185,186], suggesting the generation of autoantibodies against type I IFNs may contribute to the pathogenesis of severe COVID-19 in the late phase of infection. It would be speculated that in addition to type I IFNs, some of other cytokines such as IL-6 might also have an opposing effect in early and late phases of SARS-CoV-2 infection. Consistently, it was reported that IL-6 production levels in the early phase of RSV infection are correlated with the limitation of disease severity through its effect on maturation of regulatory T cells [187], whereas high concentrations of IL-6 in the late phase of infection correlate with respiratory failure, ARDS, and adverse clinical outcomes [188]. Thus, better understanding of molecular mechanisms underlying innate recognition-mediated immune responses in terms of immunopathology will aid to provide a therapeutic design of cytokine or anti-cytokine strategy with optimal timing and duration.
Availability of data and materials
Not applicable.
Abbreviations
- ACE2:
-
Angiotensin-converting enzyme 2
- AIM2:
-
Absence in melanoma 2
- ALR:
-
AIM2-like receptors
- AP-1:
-
Activator protein-1
- ASC:
-
Apoptosis-associated speck-like protein containing a CARD
- ASGR1:
-
Asialoglycoprotein receptor 1
- ATRA:
-
All-trans retinoic acid
- AXL:
-
Tyrosine-protein kinase receptor UFO
- BMDM:
-
Bone marrow-derived macrophage
- BST2:
-
Bone marrow stromal cell antigen 2
- CARD:
-
Caspase-recruitment domain
- cGAMP:
-
Cyclic guanosine monophosphate-adenosine monophosphate
- cGAS:
-
cGAMP synthase
- CLEC5A:
-
C-type lectin domain containing 5A
- CLEC10A:
-
C-type lectin domain containing 10A
- CLR:
-
C-type lectin receptor
- COPD:
-
Chronic obstructive pulmonary disease
- COVID-19:
-
Coronavirus disease 2019
- CTD:
-
C-terminal domain
- DAMP:
-
Damage-associated molecular pattern
- DC-SIGN:
-
Dendritic cell-specific intracellular adhesion molecule-3-grabbing nonintegrin
- E:
-
Envelop
- EMCV:
-
Encephalomyocarditis virus
- HCoV:
-
Human coronavirus
- HD:
-
Helicase domain
- HIN:
-
Hematopoietic IFN-inducible nuclear protein
- HIV:
-
Human immunodeficiency virus
- HSV-1:
-
Herpes simplex virus-1
- iE-DAP:
-
γ-D-Glutamyl-meso-diaminopimelic acid
- IFI16:
-
IFN-γ inducible protein 16
- IFN:
-
Interferon
- IL:
-
Interleukin
- IRF:
-
IFN regulatory factors
- KIM-1:
-
Kidney injury molecule-1
- LSECtin:
-
Liver and lymph node sinusoidal endothelial cell C-type lectin
- L-SIGN:
-
Liver and lymph node-specific intracellular adhesion molecule-3-grabbing nonintegrin
- LY6E:
-
Lymphocyte antigen 6 family member E
- M:
-
Membrane
- MAPK:
-
Mitogen-activated protein kinase
- MAVS:
-
Mitochondrial antiviral signaling protein
- MDA5:
-
Melanoma differentiation-associated gene 5
- MDP:
-
Muramyl dipeptide
- MERS-CoV:
-
Middle East respiratory syndrome coronavirus
- MHV:
-
Mouse hepatitis virus
- MyD88:
-
Myeloid differentiation factor 88
- N:
-
Nucleocapsid
- NF-кB:
-
Nuclear factor-кB
- NLR:
-
NOD-like receptor
- NLRP3:
-
NLR family, pyrin domain containing 3
- NOD:
-
Nucleotide-binding and oligomerization domain
- NRP1:
-
Neuropilin-1
- NSP:
-
Nonstructural protein
- ORF3a:
-
Open reading frame 3a
- PAMP:
-
Pathogen-associated molecular pattern
- PBMC:
-
Peripheral blood mononuclear cell
- RdRp:
-
RNA-dependent RNA polymerase
- PRR:
-
Pattern-recognition receptor
- RIG-I:
-
Retinoic acid-inducible gene-1
- RIPK2:
-
Recruit receptor-interacting serine/threonine-protein kinase 2
- RLR:
-
RIG-I-like receptor
- RSV:
-
Respiratory syncytial virus
- PYHIN:
-
Pyrin and HIN domain
- S:
-
Spike
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus-2
- STING:
-
Stimulator of IFN genes
- TBK1:
-
TANK-binding kinase 1
- TLR:
-
Toll-like receptor
- TMPRSS2:
-
Transmembrane protease serine 2
- TNF:
-
Tumor necrosis factor
- TRIF:
-
Toll/IL-1 receptor domain-containing adaptor inducing IFN-β
- TTYH2:
-
Tweety family member 2
- UTR:
-
Untranslated region
- 3pRNA:
-
RNAs carrying a 5′-triphosphate modification
- (−)gRNA:
-
Negative-sense RNA
- (+)gRNA:
-
Positive-sense genomic RNA.
References
Kahn JS, McIntosh K. History and recent advances in coronavirus discovery. Pediatr Infect Dis J. 2005;24(11 Suppl):S223–7.
de Vries AAF. SARS-CoV-2/COVID-19: a primer for cardiologists. Neth Heart J. 2020;28(7-8):366–83.
Drosten C, Gunther S, Preiser W, van der Werf S, Brodt HR, Becker S, et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348(20):1967–76.
Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367(19):1814–20.
Fung TS, Liu DX. Human coronavirus: host-pathogen interaction. Annu Rev Microbiol. 2019;73:529–57.
Zhang S, Wang L, Cheng G. The battle between host and SARS-CoV-2: innate immunity and viral evasion strategies. Mol Ther. 2022;30(5):1869–84.
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan. China. Lancet. 2020;395(10223):497–506.
Vabret N, Britton GJ, Gruber C, Hegde S, Kim J, Kuksin M, et al. Immunology of COVID-19: current state of the science. Immunity. 2020;52(6):910–41.
Tay MZ, Poh CM, Renia L, MacAry PA, Ng LFP. The trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol. 2020;20(6):363–74.
Li Y, Li H, Li M, Zhang L, Xie M. The prevalence, risk factors and outcome of cardiac dysfunction in hospitalized patients with COVID-19. Intensive Care Med. 2020;46(11):2096–8.
Oran DP, Topol EJ. Prevalence of asymptomatic SARS-CoV-2 infection : a narrative review. Ann Intern Med. 2020;173(5):362–7.
Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382(8):727–33.
Gandhi RT, Lynch JB, Del Rio C. Mild or moderate Covid-19. N Engl J Med. 2020;383(18):1757–66.
Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA. 2020;323(11):1061–9.
Sanchez-Ramirez DC, Mackey D. Underlying respiratory diseases, specifically COPD, and smoking are associated with severe COVID-19 outcomes: a systematic review and meta-analysis. Respir Med. 2020;171:106096.
Dessie ZG, Zewotir T. Mortality-related risk factors of COVID-19: a systematic review and meta-analysis of 42 studies and 423,117 patients. BMC Infect Dis. 2021;21(1):855.
O'Driscoll M, Ribeiro Dos Santos G, Wang L, Cummings DAT, Azman AS, Paireau J, et al. Age-specific mortality and immunity patterns of SARS-CoV-2. Nature. 2021;590(7844):140–5.
Takaoka A, Yamada T. Regulation of signaling mediated by nucleic acid sensors for innate interferon-mediated responses during viral infection. Int Immunol. 2019;31(8):477–88.
van der Made CI, Simons A, Schuurs-Hoeijmakers J, van den Heuvel G, Mantere T, Kersten S, et al. Presence of genetic variants among young men with severe COVID-19. JAMA. 2020;324(7):663–73.
Asano T, Boisson B, Onodi F, Matuozzo D, Moncada-Velez M, Maglorius Renkilaraj MRL, et al. X-linked recessive TLR7 deficiency in ~1% of men under 60 years old with life-threatening COVID-19. Sci Immunol. 2021;6(62).
Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, Chen J, et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science. 2020;370(6515):eabd4570.
Pairo-Castineira E, Clohisey S, Klaric L, Bretherick AD, Rawlik K, Pasko D, et al. Genetic mechanisms of critical illness in COVID-19. Nature. 2021;591(7848):92–8.
Del Valle DM, Kim-Schulze S, Huang HH, Beckmann ND, Nirenberg S, Wang B, et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat Med. 2020;26(10):1636–43.
Lucas C, Wong P, Klein J, Castro TBR, Silva J, Sundaram M, et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature. 2020;584(7821):463–9.
Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 2020;369(6504):718–24.
Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Moller R, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181(5):1036–45 e9.
Giamarellos-Bourboulis EJ, Netea MG, Rovina N, Akinosoglou K, Antoniadou A, Antonakos N, et al. Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe. 2020;27(6):992–1000 e3.
Zhou Z, Ren L, Zhang L, Zhong J, Xiao Y, Jia Z, et al. Heightened innate immune responses in the respiratory tract of COVID-19 patients. Cell Host Microbe. 2020;27(6):883–90 e2.
Mathew D, Giles JR, Baxter AE, Oldridge DA, Greenplate AR, Wu JE, et al. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science. 2020;369(6508):eabc8511.
Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest. 2020;130(5):2620–9.
Junqueira C, Crespo A, Ranjbar S, de Lacerda LB, Lewandrowski M, Ingber J, et al. FcgammaR-mediated SARS-CoV-2 infection of monocytes activates inflammation. Nature. 2022;606(7914):576–84.
Jackson CB, Farzan M, Chen B, Choe H. Mechanisms of SARS-CoV-2 entry into cells. Nat Rev Mol Cell Biol. 2022;23(1):3–20.
Kim D, Lee JY, Yang JS, Kim JW, Kim VN, Chang H. The architecture of SARS-CoV-2 transcriptome. Cell. 2020;181(4):914–21 e10.
Sungnak W, Huang N, Becavin C, Berg M, Queen R, Litvinukova M, et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat Med. 2020;26(5):681–7.
Ahn JH, Kim J, Hong SP, Choi SY, Yang MJ, Ju YS, et al. Nasal ciliated cells are primary targets for SARS-CoV-2 replication in the early stage of COVID-19. J Clin Invest. 2021;131(13):e148517.
Ziegler CGK, Allon SJ, Nyquist SK, Mbano IM, Miao VN, Tzouanas CN, et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell. 2020;181(5):1016–35 e19.
Hou YJ, Okuda K, Edwards CE, Martinez DR, Asakura T, Dinnon KH 3rd, et al. SARS-CoV-2 reverse genetics reveals a variable infection gradient in the respiratory tract. Cell. 2020;182(2):429–46 e14.
Domizio JD, Gulen MF, Saidoune F, Thacker VV, Yatim A, Sharma K, et al. The cGAS-STING pathway drives type I IFN immunopathology in COVID-19. Nature. 2022;603(7899):145–51.
Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–80 e8.
Zipeto D, Palmeira JDF, Arganaraz GA, Arganaraz ER. ACE2/ADAM17/TMPRSS2 interplay may be the main risk factor for COVID-19. Front Immunol. 2020;11:576745.
Gomes CP, Fernandes DE, Casimiro F, da Mata GF, Passos MT, Varela P, et al. Cathepsin L in COVID-19: from pharmacological evidences to genetics. Front Cell Infect Microbiol. 2020;10:589505.
Cantuti-Castelvetri L, Ojha R, Pedro LD, Djannatian M, Franz J, Kuivanen S, et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science. 2020;370(6518):856–60.
Lempp FA, Soriaga LB, Montiel-Ruiz M, Benigni F, Noack J, Park YJ, et al. Lectins enhance SARS-CoV-2 infection and influence neutralizing antibodies. Nature. 2021;598(7880):342–7.
Ou X, Liu Y, Lei X, Li P, Mi D, Ren L, et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun. 2020;11(1):1620.
Yang C, Zhang Y, Zeng X, Chen H, Chen Y, Yang D, et al. Kidney injury molecule-1 is a potential receptor for SARS-CoV-2. J Mol Cell Biol. 2021;13(3):185–96.
Wang S, Qiu Z, Hou Y, Deng X, Xu W, Zheng T, et al. AXL is a candidate receptor for SARS-CoV-2 that promotes infection of pulmonary and bronchial epithelial cells. Cell Res. 2021;31(2):126–40.
Wang K, Chen W, Zhang Z, Deng Y, Lian JQ, Du P, et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct Target Ther. 2020;5(1):283.
Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, et al. Structure of M (pro) from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582(7811):289–93.
Hillen HS, Kokic G, Farnung L, Dienemann C, Tegunov D, Cramer P. Structure of replicating SARS-CoV-2 polymerase. Nature. 2020;584(7819):154–6.
Zhao Y, Sun J, Li Y, Li Z, Xie Y, Feng R, et al. The strand-biased transcription of SARS-CoV-2 and unbalanced inhibition by remdesivir. iScience. 2021;24(8):102857.
Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol. 2015;33:257–90.
Kanneganti TD. Intracellular innate immune receptors: life inside the cell. Immunol Rev. 2020;297(1):5–12.
Goubau D, Deddouche S. Reis e Sousa C. Cytosolic sensing of viruses. Immunity. 2013;38(5):855–69.
Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–20.
Jung S, Potapov I, Chillara S, Del Sol A. Leveraging systems biology for predicting modulators of inflammation in patients with COVID-19. Sci Adv. 2021;7(6):eabe5735.
Zheng M, Karki R, Williams EP, Yang D, Fitzpatrick E, Vogel P, et al. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat Immunol. 2021;22(7):829–38.
Khan S, Shafiei MS, Longoria C, Schoggins JW, Savani RC, Zaki H. SARS-CoV-2 spike protein induces inflammation via TLR2-dependent activation of the NF-kappaB pathway. Elife. 2021;10:e68563.
Bortolotti D, Gentili V, Rizzo S, Schiuma G, Beltrami S, Strazzabosco G, et al. TLR3 and TLR7 RNA sensor activation during SARS-CoV-2 infection. Microorganisms. 2021;9(9):1820.
Zhao Y, Kuang M, Li J, Zhu L, Jia Z, Guo X, et al. SARS-CoV-2 spike protein interacts with and activates TLR41. Cell Res. 2021;31(7):818–20.
Shirato K, Kizaki T. SARS-CoV-2 spike protein S1 subunit induces pro-inflammatory responses via toll-like receptor 4 signaling in murine and human macrophages. Heliyon. 2021;7(2):e06187.
Choudhury A, Mukherjee S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J Med Virol. 2020;92(10):2105–13.
Yin X, Riva L, Pu Y, Martin-Sancho L, Kanamune J, Yamamoto Y, et al. MDA5 governs the innate immune response to SARS-CoV-2 in lung epithelial cells. Cell Rep. 2021;34(2):108628.
Rebendenne A, Valadao ALC, Tauziet M, Maarifi G, Bonaventure B, McKellar J, et al. SARS-CoV-2 triggers an MDA-5-dependent interferon response which is unable to control replication in lung epithelial cells. J Virol. 2021;95(8):e02415-20.
Sampaio NG, Chauveau L, Hertzog J, Bridgeman A, Fowler G, Moonen JP, et al. The RNA sensor MDA5 detects SARS-CoV-2 infection. Sci Rep. 2021;11(1):13638.
Yang DM, Geng TT, Harrison AG, Wang PH. Differential roles of RIG-I like receptors in SARS-CoV-2 infection. Mil Med Res. 2021;8(1):49.
Thorne LG, Reuschl AK, Zuliani-Alvarez L, Whelan MVX, Turner J, Noursadeghi M, et al. SARS-CoV-2 sensing by RIG-I and MDA5 links epithelial infection to macrophage inflammation. EMBO J. 2021;40(15):e107826.
Yamada T, Sato S, Sotoyama Y, Orba Y, Sawa H, Yamauchi H, et al. RIG-I triggers a signaling-abortive anti-SARS-CoV-2 defense in human lung cells. Nat Immunol. 2021;22(7):820–8.
Kouwaki T, Nishimura T, Wang G, Oshiumi H. RIG-I-like receptor-mediated recognition of viral genomic RNA of severe acute respiratory syndrome coronavirus-2 and viral escape from the host innate immune responses. Front Immunol. 2021;12:700926.
Lu Q, Liu J, Zhao S, Gomez Castro MF, Laurent-Rolle M, Dong J, et al. SARS-CoV-2 exacerbates proinflammatory responses in myeloid cells through C-type lectin receptors and Tweety family member 2. Immunity. 2021;54(6):1304–19 e9.
Rodrigues TS, de Sa KSG, Ishimoto AY, Becerra A, Oliveira S, Almeida L, et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J Exp Med. 2021;218(3):e20201707.
Ferreira AC, Soares VC, de Azevedo-Quintanilha IG, Dias S, Fintelman-Rodrigues N, Sacramento CQ, et al. SARS-CoV-2 engages inflammasome and pyroptosis in human primary monocytes. Cell Death Discov. 2021;7(1):43.
Campbell GR, To RK, Hanna J, Spector SA. SARS-CoV-2, SARS-CoV-1, and HIV-1 derived ssRNA sequences activate the NLRP3 inflammasome in human macrophages through a non-classical pathway. iScience. 2021;24(4):102295.
Xu H, Akinyemi IA, Chitre SA, Loeb JC, Lednicky JA, McIntosh MT, et al. SARS-CoV-2 viroporin encoded by ORF3a triggers the NLRP3 inflammatory pathway. Virology. 2022;568:13–22.
Pan P, Shen M, Yu Z, Ge W, Chen K, Tian M, et al. SARS-CoV-2 N protein promotes NLRP3 inflammasome activation to induce hyperinflammation. Nat Commun. 2021;12(1):4664.
Theobald SJ, Simonis A, Georgomanolis T, Kreer C, Zehner M, Eisfeld HS, et al. Long-lived macrophage reprogramming drives spike protein-mediated inflammasome activation in COVID-19. EMBO Mol Med. 2021;13(8):e14150.
Neufeldt CJ, Cerikan B, Cortese M, Frankish J, Lee JY, Plociennikowska A, et al. SARS-CoV-2 infection induces a pro-inflammatory cytokine response through cGAS-STING and NF-kappaB. Commun Biol. 2022;5(1):45.
Liu X, Wei L, Xu F, Zhao F, Huang Y, Fan Z, et al. SARS-CoV-2 spike protein-induced cell fusion activates the cGAS-STING pathway and the interferon response. Sci Signal. 2022;15(729):eabg8744.
Povysil G, Butler-Laporte G, Shang N, Wang C, Khan A, Alaamery M, et al. Rare loss-of-function variants in type I IFN immunity genes are not associated with severe COVID-19. J Clin Invest. 2021;131(14):e147834.
Ma J, Zhu F, Zhao M, Shao F, Yu D, Ma J, et al. SARS-CoV-2 nucleocapsid suppresses host pyroptosis by blocking Gasdermin D cleavage. EMBO J. 2021;40(18):e108249.
Fitzgerald KA, Kagan JC. Toll-like receptors and the control of immunity. Cell. 2020;180(6):1044–66.
Liu G, Zhao Y. Toll-like receptors and immune regulation: their direct and indirect modulation on regulatory CD4+ CD25+ T cells. Immunology. 2007;122(2):149–56.
De Nardo D. Toll-like receptors: activation, signalling and transcriptional modulation. Cytokine. 2015;74(2):181–9.
Ariza ME, Glaser R, Kaumaya PT, Jones C, Williams MV. The EBV-encoded dUTPase activates NF-kappa B through the TLR2 and MyD88-dependent signaling pathway. J Immunol. 2009;182(2):851–9.
Cooper A, Tal G, Lider O, Shaul Y. Cytokine induction by the hepatitis B virus capsid in macrophages is facilitated by membrane heparan sulfate and involves TLR2. J Immunol. 2005;175(5):3165–76.
Cai M, Li M, Wang K, Wang S, Lu Q, Yan J, et al. The herpes simplex virus 1-encoded envelope glycoprotein B activates NF-kappaB through the Toll-like receptor 2 and MyD88/TRAF6-dependent signaling pathway. PLoS One. 2013;8(1):e54586.
Kurt-Jones EA, Popova L, Kwinn L, Haynes LM, Jones LP, Tripp RA, et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol. 2000;1(5):398–401.
Georgel P, Jiang Z, Kunz S, Janssen E, Mols J, Hoebe K, et al. Vesicular stomatitis virus glycoprotein G activates a specific antiviral Toll-like receptor 4-dependent pathway. Virology. 2007;362(2):304–13.
Chao CH, Wu WC, Lai YC, Tsai PJ, Perng GC, Lin YS, et al. Dengue virus nonstructural protein 1 activates platelets via Toll-like receptor 4, leading to thrombocytopenia and hemorrhage. PLoS Pathog. 2019;15(4):e1007625.
Lai CY, Strange DP, Wong TAS, Lehrer AT, Verma S. Ebola virus glycoprotein induces an innate immune response in vivo via TLR4. Front Microbiol. 2017;8:1571.
Totura AL, Whitmore A, Agnihothram S, Schafer A, Katze MG, Heise MT, et al. Toll-like receptor 3 signaling via TRIF contributes to a protective innate immune response to severe acute respiratory syndrome coronavirus infection. mBio. 2015;6(3):e00638–15.
Cervantes-Barragan L, Zust R, Weber F, Spiegel M, Lang KS, Akira S, et al. Control of coronavirus infection through plasmacytoid dendritic-cell-derived type I interferon. Blood. 2007;109(3):1131–7.
Scheuplein VA, Seifried J, Malczyk AH, Miller L, Hocker L, Vergara-Alert J, et al. High secretion of interferons by human plasmacytoid dendritic cells upon recognition of Middle East respiratory syndrome coronavirus. J Virol. 2015;89(7):3859–69.
Sohn KM, Lee SG, Kim HJ, Cheon S, Jeong H, Lee J, et al. COVID-19 patients upregulate Toll-like receptor 4-mediated inflammatory signaling that mimics bacterial sepsis. J Korean Med Sci. 2020;35(38):e343.
Rehwinkel J, Reis e Sousa C. RIGorous detection: exposing virus through RNA sensing. Science. 2010;327(5963):284–6.
Kell AM, Gale M Jr. RIG-I in RNA virus recognition. Virology. 2015;479-480:110–21.
Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441(7089):101–5.
Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, et al. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med. 2008;205(7):1601–10.
Dias Junior AG, Sampaio NG, Rehwinkel J. A balancing act: MDA5 in antiviral immunity and autoinflammation. Trends Microbiol. 2019;27(1):75–85.
Li J, Liu Y, Zhang X. Murine coronavirus induces type I interferon in oligodendrocytes through recognition by RIG-I and MDA5. J Virol. 2010;84(13):6472–82.
Zalinger ZB, Elliott R, Rose KM, Weiss SR. MDA5 is critical to host defense during infection with murine coronavirus. J Virol. 2015;89(24):12330–40.
Roth-Cross JK, Bender SJ, Weiss SR. Murine coronavirus mouse hepatitis virus is recognized by MDA5 and induces type I interferon in brain macrophages/microglia. J Virol. 2008;82(20):9829–38.
Zhao X, Chu H, Wong BH, Chiu MC, Wang D, Li C, et al. Activation of C-type lectin receptor and (RIG)-I-like receptors contributes to proinflammatory response in Middle East respiratory syndrome coronavirus-infected macrophages. J Infect Dis. 2020;221(4):647–59.
Loske J, Rohmel J, Lukassen S, Stricker S, Magalhaes VG, Liebig J, et al. Pre-activated antiviral innate immunity in the upper airways controls early SARS-CoV-2 infection in children. Nat Biotechnol. 2022;40(3):319–24.
Zhao Q, Meng M, Kumar R, Wu Y, Huang J, Lian N, et al. The impact of COPD and smoking history on the severity of COVID-19: a systemic review and meta-analysis. J Med Virol. 2020;92(10):1915–21.
Attaway AA, Zein J, Hatipoglu US. SARS-CoV-2 infection in the COPD population is associated with increased healthcare utilization: an analysis of Cleveland clinic’s COVID-19 registry. EClinicalMedicine. 2020;26:100515.
Sin DD. COVID-19 in COPD: a growing concern. EClinicalMedicine. 2020;26:100546.
Liu TX, Zhang JW, Tao J, Zhang RB, Zhang QH, Zhao CJ, et al. Gene expression networks underlying retinoic acid-induced differentiation of acute promyelocytic leukemia cells. Blood. 2000;96(4):1496–504.
Geijtenbeek TB, Gringhuis SI. Signalling through C-type lectin receptors: shaping immune responses. Nat Rev Immunol. 2009;9(7):465–79.
Bermejo-Jambrina M, Eder J, Helgers LC, Hertoghs N, Nijmeijer BM, Stunnenberg M, et al. C-type lectin receptors in antiviral immunity and viral escape. Front Immunol. 2018;9:590.
Zhao D, Han X, Zheng X, Wang H, Yang Z, Liu D, et al. The myeloid LSECtin is a DAP12-coupled receptor that is crucial for inflammatory response induced by Ebola virus glycoprotein. PLoS Pathog. 2016;12(3):e1005487.
Chen ST, Lin YL, Huang MT, Wu MF, Cheng SC, Lei HY, et al. CLEC5A is critical for dengue-virus-induced lethal disease. Nature. 2008;453(7195):672–6.
Wu MF, Chen ST, Yang AH, Lin WW, Lin YL, Chen NJ, et al. CLEC5A is critical for dengue virus-induced inflammasome activation in human macrophages. Blood. 2013;121(1):95–106.
Marzi A, Gramberg T, Simmons G, Moller P, Rennekamp AJ, Krumbiegel M, et al. DC-SIGN and DC-SIGNR interact with the glycoprotein of Marburg virus and the S protein of severe acute respiratory syndrome coronavirus. J Virol. 2004;78(21):12090–5.
Pohlmann S, Baribaud F, Lee B, Leslie GJ, Sanchez MD, Hiebenthal-Millow K, et al. DC-SIGN interactions with human immunodeficiency virus type 1 and 2 and simian immunodeficiency virus. J Virol. 2001;75(10):4664–72.
Hillaire ML, Nieuwkoop NJ, Boon AC, de Mutsert G, Vogelzang-van Trierum SE, Fouchier RA, et al. Binding of DC-SIGN to the hemagglutinin of influenza A viruses supports virus replication in DC-SIGN expressing cells. PLoS One. 2013;8(2):e56164.
Tassaneetrithep B, Burgess TH, Granelli-Piperno A, Trumpfheller C, Finke J, Sun W, et al. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J Exp Med. 2003;197(7):823–9.
de Witte L, Abt M, Schneider-Schaulies S, van Kooyk Y, Geijtenbeek TB. Measles virus targets DC-SIGN to enhance dendritic cell infection. J Virol. 2006;80(7):3477–86.
Alvarez CP, Lasala F, Carrillo J, Muniz O, Corbi AL, Delgado R. C-type lectins DC-SIGN and L-SIGN mediate cellular entry by Ebola virus in cis and in trans. J Virol. 2002;76(13):6841–4.
Jeffers SA, Tusell SM, Gillim-Ross L, Hemmila EM, Achenbach JE, Babcock GJ, et al. CD209L (L-SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proc Natl Acad Sci U S A. 2004;101(44):15748–53.
Brudner M, Karpel M, Lear C, Chen L, Yantosca LM, Scully C, et al. Lectin-dependent enhancement of Ebola virus infection via soluble and transmembrane C-type lectin receptors. PLoS One. 2013;8(4):e60838.
Thepaut M, Luczkowiak J, Vives C, Labiod N, Bally I, Lasala F, et al. DC/L-SIGN recognition of spike glycoprotein promotes SARS-CoV-2 trans-infection and can be inhibited by a glycomimetic antagonist. PLoS Pathog. 2021;17(5):e1009576.
Amraei R, Yin W, Napoleon MA, Suder EL, Berrigan J, Zhao Q, et al. CD209L/L-SIGN and CD209/DC-SIGN act as receptors for SARS-CoV-2. ACS Cent Sci. 2021;7(7):1156–65.
Inohara C, McDonald C, Nunez G. NOD-LRR proteins: role in host-microbial interactions and inflammatory disease. Annu Rev Biochem. 2005;74:355–83.
Zhong Y, Kinio A, Saleh M. Functions of NOD-like receptors in human diseases. Front Immunol. 2013;4:333.
Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417–26.
Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. 2004;20(3):319–25.
Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19(8):477–89.
Guarda G, Zenger M, Yazdi AS, Schroder K, Ferrero I, Menu P, et al. Differential expression of NLRP3 among hematopoietic cells. J Immunol. 2011;186(4):2529–34.
Chen IY, Moriyama M, Chang MF, Ichinohe T. Severe acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome. Front Microbiol. 2019;10:50.
Nieto-Torres JL, Verdia-Baguena C, Jimenez-Guardeno JM, Regla-Nava JA, Castano-Rodriguez C, Fernandez-Delgado R, et al. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology. 2015;485:330–9.
Shi CS, Nabar NR, Huang NN, Kehrl JH. SARS-coronavirus open reading frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. 2019;5:101.
Castano-Rodriguez C, Honrubia JM, Gutierrez-Alvarez J, DeDiego ML, Nieto-Torres JL, Jimenez-Guardeno JM, et al. Role of severe acute respiratory syndrome coronavirus viroporins E, 3a, and 8a in Replication and Pathogenesis. mBio. 2018;9(3):e02325-17.
Ito M, Yanagi Y, Ichinohe T. Encephalomyocarditis virus viroporin 2B activates NLRP3 inflammasome. PLoS Pathog. 2012;8(8):e1002857.
Ichinohe T, Pang IK, Iwasaki A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat Immunol. 2010;11(5):404–10.
Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity. 2009;30(4):556–65.
Thomas PG, Dash P, Aldridge JR Jr, Ellebedy AH, Reynolds C, Funk AJ, et al. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity. 2009;30(4):566–75.
Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med. 2009;206(1):79–87.
Caruso R, Warner N, Inohara N, Nunez G. NOD1 and NOD2: signaling, host defense, and inflammatory disease. Immunity. 2014;41(6):898–908.
Fan YH, Roy S, Mukhopadhyay R, Kapoor A, Duggal P, Wojcik GL, et al. Role of nucleotide-binding oligomerization domain 1 (NOD1) and its variants in human Cytomegalovirus control in vitro and in vivo. Proc Natl Acad Sci U S A. 2016;113(48):E7818–E27.
Vegna S, Gregoire D, Moreau M, Lassus P, Durantel D, Assenat E, et al. NOD1 participates in the innate immune response triggered by hepatitis C virus polymerase. J Virol. 2016;90(13):6022–35.
Sabbah A, Chang TH, Harnack R, Frohlich V, Tominaga K, Dube PH, et al. Activation of innate immune antiviral responses by Nod2. Nat Immunol. 2009;10(10):1073–80.
Kapoor A, Forman M, Arav-Boger R. Activation of nucleotide oligomerization domain 2 (NOD2) by human Cytomegalovirus initiates innate immune responses and restricts virus replication. PLoS One. 2014;9(3):e92704.
Lupfer C, Thomas PG, Kanneganti TD. Nucleotide oligomerization and binding domain 2-dependent dendritic cell activation is necessary for innate immunity and optimal CD8+ T cell responses to influenza A virus infection. J Virol. 2014;88(16):8946–55.
Wu XM, Zhang J, Li PW, Hu YW, Cao L, Ouyang S, et al. NOD1 promotes antiviral signaling by binding viral RNA and regulating the interaction of MDA5 and MAVS. J Immunol. 2020;204(8):2216–31.
Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458(7237):509–13.
Burckstummer T, Baumann C, Bluml S, Dixit E, Durnberger G, Jahn H, et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol. 2009;10(3):266–72.
Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol. 2010;11(11):997–1004.
Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol. 2010;11(5):395–402.
Zhang H, Luo J, Alcorn JF, Chen K, Fan S, Pilewski J, et al. AIM2 inflammasome is critical for influenza-induced lung injury and mortality. J Immunol. 2017;198(11):4383–93.
Strittmatter GE, Sand J, Sauter M, Seyffert M, Steigerwald R, Fraefel C, et al. IFN-gamma primes keratinocytes for HSV-1-induced inflammasome activation. J Invest Dermatol. 2016;136(3):610–20.
Connolly DJ, Bowie AG. The emerging role of human PYHIN proteins in innate immunity: implications for health and disease. Biochem Pharmacol. 2014;92(3):405–14.
Briard B, Place DE, Kanneganti TD. DNA sensing in the innate immune response. Physiology (Bethesda). 2020;35(2):112–24.
Moriyama M, Nagai M, Maruzuru Y, Koshiba T, Kawaguchi Y, Ichinohe T. Influenza virus-induced oxidized DNA activates inflammasomes. iScience. 2020;23(7):101270.
Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun. 2019;10(1):1689.
Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339(6121):786–91.
Wu J, Sun L, Chen X, Du F, Shi H, Chen C, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339(6121):826–30.
Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Rohl I, et al. cGAS produces a 2'-5'-linked cyclic dinucleotide second messenger that activates STING. Nature. 2013;498(7454):380–4.
Hopfner KP, Hornung V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat Rev Mol Cell Biol. 2020;21(9):501–21.
West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520(7548):553–7.
Rongvaux A, Jackson R, Harman CC, Li T, West AP, de Zoete MR, et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell. 2014;159(7):1563–77.
Huang LS, Hong Z, Wu W, Xiong S, Zhong M, Gao X, et al. mtDNA activates cGAS signaling and suppresses the YAP-mediated endothelial cell proliferation program to promote inflammatory injury. Immunity. 2020;52(3):475–86 e5.
Jiang H, Xue X, Panda S, Kawale A, Hooy RM, Liang F, et al. Chromatin-bound cGAS is an inhibitor of DNA repair and hence accelerates genome destabilization and cell death. EMBO J. 2019;38(21):e102718.
Riley JS, Quarato G, Cloix C, Lopez J, O'Prey J, Pearson M, et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J 2018, 37(17) e99238
White MJ, McArthur K, Metcalf D, Lane RM, Cambier JC, Herold MJ, et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell. 2014;159(7):1549–62.
Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. 2017;548(7668):461–5.
Aarreberg LD, Esser-Nobis K, Driscoll C, Shuvarikov A, Roby JA, Gale M Jr. Interleukin-1beta induces mtDNA release to activate innate immune signaling via cGAS-STING. Mol Cell. 2019;74(4):801–15 e6.
Maekawa H, Inoue T, Ouchi H, Jao TM, Inoue R, Nishi H, et al. Mitochondrial damage causes inflammation via cGAS-STING signaling in acute kidney injury. Cell Rep. 2019;29(5):1261–73 e6.
Hartlova A, Erttmann SF, Raffi FA, Schmalz AM, Resch U, Anugula S, et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity. 2015;42(2):332–43.
Mackenzie KJ, Carroll P, Lettice L, Tarnauskaite Z, Reddy K, Dix F, et al. Ribonuclease H2 mutations induce a cGAS/STING-dependent innate immune response. EMBO J. 2016;35(8):831–44.
Gao D, Li T, Li XD, Chen X, Li QZ, Wight-Carter M, et al. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc Natl Acad Sci U S A. 2015;112(42):E5699–705.
Aguirre S, Luthra P, Sanchez-Aparicio MT, Maestre AM, Patel J, Lamothe F, et al. Dengue virus NS2B protein targets cGAS for degradation and prevents mitochondrial DNA sensing during infection. Nat Microbiol. 2017;2:17037.
Li XD, Wu J, Gao D, Wang H, Sun L, Chen ZJ. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science. 2013;341(6152):1390–4.
Paijo J, Doring M, Spanier J, Grabski E, Nooruzzaman M, Schmidt T, et al. cGAS senses human Cytomegalovirus and induces type i interferon responses in human monocyte-derived cells. PLoS Pathog. 2016;12(4):e1005546.
Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature. 2014;505(7485):691–5.
Andreakos E. STINGing type I IFN-mediated immunopathology in COVID-19. Nat Immunol. 2022;23(4):478–80.
Li H, Zhou F, Zhang L. STING, a critical contributor to SARS-CoV-2 immunopathology. Signal Transduct Target Ther. 2022;7(1):106.
Pfaender S, Mar KB, Michailidis E, Kratzel A, Boys IN, V'Kovski P, et al. LY6E impairs coronavirus fusion and confers immune control of viral disease. Nat Microbiol. 2020;5(11):1330–9.
Martin-Sancho L, Lewinski MK, Pache L, Stoneham CA, Yin X, Becker ME, et al. Functional landscape of SARS-CoV-2 cellular restriction. Mol Cell. 2021;81(12):2656–68 e8.
Zhou Q, Chen V, Shannon CP, Wei XS, Xiang X, Wang X, et al. Interferon-alpha2b treatment for COVID-19. Front Immunol. 2020;11:1061.
Wang N, Zhan Y, Zhu L, Hou Z, Liu F, Song P, et al. Retrospective multicenter cohort study shows early interferon therapy is associated with favorable clinical responses in COVID-19 patients. Cell Host Microbe. 2020;28(3):455–64 e2.
Levy R, Bastard P, Lanternier F, Lecuit M, Zhang SY, Casanova JL. IFN-alpha2a therapy in two patients with inborn errors of TLR3 and IRF3 infected with SARS-CoV-2. J Clin Immunol. 2021;41(1):26–7.
Feld JJ, Kandel C, Biondi MJ, Kozak RA, Zahoor MA, Lemieux C, et al. Peginterferon lambda for the treatment of outpatients with COVID-19: a phase 2, placebo-controlled randomised trial. Lancet Respir Med. 2021;9(5):498–510.
Major J, Crotta S, Llorian M, McCabe TM, Gad HH, Priestnall SL, et al. Type I and III interferons disrupt lung epithelial repair during recovery from viral infection. Science. 2020;369(6504):712–7.
Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann HH, Zhang Y, et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science. 2020;370(6515):eabd4585.
Lopez J, Mommert M, Mouton W, Pizzorno A, Brengel-Pesce K, Mezidi M, et al. Early nasal type I IFN immunity against SARS-CoV-2 is compromised in patients with autoantibodies against type I IFNs. J Exp Med. 2021;218(10):e20211211.
Bastard P, Gervais A, Le Voyer T, Rosain J, Philippot Q, Manry J, et al. Autoantibodies neutralizing type I IFNs are present in ~4% of uninfected individuals over 70 years old and account for ~20% of COVID-19 deaths. Sci Immunol. 2021;6(62):eabl4340.
Pyle CJ, Uwadiae FI, Swieboda DP, Harker JA. PLoS Pathog. 2017;13(9):e1006640.
Moore JB, June CH. Science. 2020;368(6490):473–4.
Acknowledgements
We thank the other collaborators, Yasuko Orba, Hirofumi Sawa, and Michihito Sasaki, who contributed to our work shown in this review.
Funding
Our work shown in this review was supported by grants from the Japan Society for the Promotion of Science (JSPS; Grant-in-Aid for Challenging Research (Pioneering), JP17H06265, to A.T.); Grant-in-Aid for Young Scientists (B; JP19K16664, to T. Y.); Japan Agency for Medical Research and Development (AMED; Program on the Innovative Development and the Application of New Drugs for Hepatitis B, JP18fk0310101 (to A. T.)); the Naito Foundation and Asai Germanium Research Institute (to A. T.); Akiyama Life Science Foundation (to A. T.); and the Uehara Memorial Foundation (to A. T.).
Author information
Authors and Affiliations
Contributions
TY and AT wrote the manuscript. The authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yamada, T., Takaoka, A. Innate immune recognition against SARS-CoV-2. Inflamm Regener 43, 7 (2023). https://doi.org/10.1186/s41232-023-00259-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41232-023-00259-5