
Abstract
Aim
This study aimed to develop a multiplex PCR assay for simultaneous detection of major Gram-negative etiologies of septicemia and evaluate its performance.
Methods
Multiplex PCR (mPCR) assays were developed targeting 11 bacterial strains. Species-specific primers were confirmed using known clinical isolates and standard strains. Gradient PCR was performed on each primer against its target bacterial gene to determine its optimal amplification condition. The minimum detectable DNA concentration of the two assays was evaluated by adjusting bacterial DNA concentration to 100 ng/μL and, tenfold serially diluting it up to 10 pg/μL with DNAse-free water. The diagnostic accuracy of mPCR assays was established by subjecting the assays to 60 clinical blood samples.
Results
Two mPCR assays were developed. Optimal primer annealing temperature of 55 °C was established and utilized in the final amplification conditions. The assays detected all targeted bacteria, with a 100 pg minimum detectable DNA concentration. Pathogens were not detected directly from whole blood, but after 4 h and 8 h of incubation, 41% (5/12) and 100% (12/12) of the bacteria were detected in culture fluids, respectively. The assays also identified Salmonella spp. and Klebsiella pneumoniae co-infections and extra pathogens (1 E. coli and 2 K. pneumoniae) compared with culture. The sensitivity and specificity of the mPCR were 100.0% (71.7–100.0) and 98.0% (90.7–99.0), respectively. The area under the ROC curve was 1.00 (1.00–1.00).
Conclusions
The mPCR assays demonstrated substantial potential as a rapid tool for septicemia diagnosis alongside the traditional blood culture method. Notably, it was able to identify additional isolates, detect co-infections, and efficiently detect low bacterial DNA loads with high sensitivity, implying its value in enhancing efficiency of diagnosis of septicemia.
Similar content being viewed by others


Introduction
Septicemia is an invasion of the bloodstream by bacteria originating from other body organs that remains a threat to public health worldwide [1]. Although the burden of septicemia is not well-elucidated, prevalence in most low-income settings ranges between 2 and 15% [2,3,4,5]. The vast majority of morbidity and mortality due to septicemia occur in low-and-middle-income countries [4, 6], with malnutrition and poor health infrastructure increasing susceptibility [7]. Children below 5 years and the elderly are disproportionately affected [8]. Even though advancements in treatment and critical care have substantially improved the management of septicemia, morbidity is still high in most high-burden countries, and global under-five mortality targets due to septicemia are not yet achieved [6]. Inadequate sensitivity of available diagnostic approaches makes early detection extremely difficult and largely accounts for undesirable outcomes. Delays in initiation of treatment increases the likelihood of mortality and may have serious consequences [9], especially in neonates [10]. A rapid and sensitive diagnostic test is, therefore, a priority, not only to improve patient outcomes but also to achieve antimicrobial stewardship.
Etiologies of septicemia include both Gram-positive bacteria (GPB) and Gram-negative bacteria (GNB) [11, 12], epidemiologically differing by region, gender and age [13, 14]. GNB, including Klebsiella pneumoniae, Escherichia coli, Proteus spp., Pseudomonas spp., Enterobacter spp., Citrobacter spp., and Acinetobacter spp., have been identified as the most frequent causes of septicemia, particularly among neonates and younger children [15, 16]. Most importantly, they are increasingly associated with septic shock [15, 17], with an elevated likelihood of developing antibiotic resistance [19], and generally poor clinical outcomes [20]. Therefore, timely and accurate diagnosis is critical for improved patient management.
Diagnosis of septicemia mainly depends on detecting causative microorganisms in a blood sample through blood culture, which usually takes up to 5 days for potential causative organisms to grow, be identified and profile their sensitivity to antibiotics. Given the importance of early and adequate initiation of antibiotic therapy for patients with septicemia, blood culture falls short of timeliness, a crucial element of an ideal diagnostic test. Moreover, its accuracy is affected by fastidious microorganisms, contaminants or prior exposure to antibiotics [11]. Furthermore, inadequate blood sample volume coupled with the transient or intermittent nature of septicemia can complicate diagnosis in children [12].
Several novel technologies for septicemia diagnosis, based on molecular techniques of detecting pathogens at strain level, have been developed to circumvent blood culture shortfalls [13]. Although these tools have high initial and operation costs, their potential to address gaps in pathogen identification and susceptibility to antibiotics makes them a viable alternative to conventional methods. Initial technologies were predominantly singleplex PCR-based tools, whose main shortfall was their inability to identify co-infections, given the polymicrobial nature of septicemia [14]. Consequently, several multiplex PCR assays have been developed [15], with the advantage of broad-range microbial detection, that are more suitable for the clinical setting. However, the broad-range target compromises their sensitivity [16, 17], and the panels may not contain all clinically relevant pathogens.
This study developed two sets of mPCR assays to detect 11 (Table 1) select GNB etiologies of septicemia in children. The 11 organisms were selected based on unpublished data from ongoing surveillance within the study area, and other studies in Kenya [18, 19] and in the region [20, 21] that have also frequently observed these organisms among children with septicemia.
Materials and methods
Bacterial strains
Eleven target standard reference strains purchased from the American Type Culture Collection [American type culture collection (ATCC)] (Manassas, VA, USA) and five off-target strains (Table 1) were selected for confirmation of species primers. The off-target strains were well-characterized clinical isolates from Kiambu and Homabay Counties in Kenya that were stored at − 80 °C in the NUITM–KEMRI Project, Kenya Research Station. Target bacterial stocked strains (Table 2) for confirming species-specific primers were also identified.
To revive the strains, the stocked isolates were transferred from − 80 °C into ice. A loopful of glycerol stock was streaked on LB-Agar medium, and incubated at 37 °C for 18–24 h. The identity of all isolates using the automated identification system VITEK-2 GN cards (Biomeriux, France) and monoplex PCR.
Primer selection and design
We used ten sets of primers (Table 3), of which six were previously published sets targeting known species-specific genes. Remaining four sets were designed for this study based on the conserved regions, using online primer design software (https://bioinfo.ut.ee/primer3/). The study utilized primers with no analytical cross-reactivity, according to in silico multiple sequence alignment in MPprimer software [22], and synthesized by Sigma (Sigma-Aldrich, Germany).
Bacterial DNA extraction
Bacterial DNA from clinical stocked and standard strains in 250 µL broth Luria–Bertani (LB) were extracted using the QIAamp DNA Kit (Qiagen, Netherlands), following the manufacturer’s instructions. The resultant DNA quantity and purity were determined using Qubit and NanoDrop 2000, (Thermo Scientific, USA) spectrophotometers.
Development of mPCR assays
Initially, this study determined the primer’s annealing temperature by running monoplex PCR assays for each primer pair using the gradient PCR approach, which were ranging between 52 and 58 °C. Assays were of 25 μL reaction volume, comprising of Illustra™ Ready-To-Go™ Beads (GE Healthcare-life science), molecular grade water (22 μL), DNA template (2 μL), and specific primer (1 μL) of 10 pmol/μL working concentration solution. PCR amplifications were performed using Biorad-iCycleriQ thermocycler (Bio-Rad, USA) with the following conditions: initial denaturation at 95 °C for 3 min; 35 cycles of denaturation at 95 °C for 30 s, annealing at 52–58 °C for 45 s and extension at 72 °C for 1 min, and final extension at 72 °C for 7 min. PCR products were observed by minigel electrophoresis using 2% agarose gel with Tris Borate EDTA (TBE) buffer at 100 V for 30 min.
For optimizing the mPCR assays, two specific primer pairs were mixed, amplified and their product sizes evaluated. A primer set was added consecutively until all other primer sets were mixed, and optimal amplification confirmed. Amplification conditions were the same as those of monoplex assays, but the concentration of a primer pair used at each stage was adjusted based on the previous primer pair product. To minimize overcrowding of PCR product bands and to increase the accuracy of the mPCR, the specific targets were grouped into two sets as follows: Salmonella spp., Escherichia coli, Shigella spp., Acinetobacter baumannii, Pseudomonas aeruginosa, and Vibrio cholerae in mPCR A assay and Campylobacter spp., Enterobacter spp., Aeromonas spp., Providentia alcalifaciens, and Klebsiella pneumoniae in mPCR B assay. Results of these the two mPCR assays were confirmed by running singleplex PCRs using single primer sets.
Analytical performance of the two mPCR assays
To evaluate the limit of detection (LOD) of the mPCR assays, the concentration of DNA template for each target reference strain was adjusted to 100 ng/μL and tenfolds serially diluted up to 10 pg/μL using DNAse-free water. The serial dilution of each target DNA template was then amplified by the optimized mPCR reaction system. To evaluate the reproducibility of the assays, each serial dilution was prepared in triplicates. mPCR inter-assay was performed on each dilution.
Performance of the multiplex assays on clinical samples
Performance of the mPCR assays was evaluated by subjecting them to clinical venous blood samples collected from 60 children aged below 5 years. These were children presenting with septicemia symptomatically (at least two of the following signs or symptoms, including fever (temp > 38 °C or < 36 °C), WBC > 12,000 cells/mm3, or < 4000 cells/mm3, or bands > 10%, respiratory Rate > 24 breaths/min, and Heart Rate > 90 beats/min) at Kiambu County Referral Hospital, Kenya. Approximately 8 mL of venous blood was collected from each participant. One ml aliquot of blood was put in an EDTA tube and stored at 4 °C for bacterial DNA extraction for mPCR assays, and 3 mL of blood put in duplicates in a biphasic blood culture medium (Himedia, India) and immediately transported to the NUITM laboratory for analysis.
The biphasic blood cultures were incubated aerobically at 37 °C and microaerophilically at 42 °C for up to 5 days, with subculture after every 24 h on MacConkey’s agar (Himedia, India), Xylose Lysine Deoxycholate (XLD) agar (Himedia, India), Thiosulfate-Citrate-Bile Salts-Sucrose (TCBS) agar (Eiken Chemicals, Japan) for V. cholerae, and Blood agar base (Oxoid, UK) with campylobacter selective supplement (Oxoid, UK) and 10% defibrinated sheep blood for Campylobacter spp. The subcultures were incubated for 24 h aerobically at 37 °C, except for blood agar for Campylobacter spp., which was done micro-aerophilically at 42 °C for the same duration. Resultant colonies were identified by Gram staining and Vitek 2 GN automatic cards identification system (Biomérieux, France).
About 0.5 mL sample culture broth was collected into 1 mL cryo-vial after every 4 h of incubation up to 20 h for DNA extraction to determine minimum detection time; the earliest timepoint the newly developed mPCR would detect target bacterial DNA in a blood culture before recovery of the isolate in subculture media. Bacterial DNA was extracted from culture broth and whole blood samples using the QIAamp DNA Blood Mini Kit (Qiagen, Netherlands), with 200 µL of the two sample types separately aliquoted into 2.0 mL sample tubes containing proteinase K on an ice block and following the manufacturer’s instructions. Before PCR amplification, Qubit™ fluorometer (Thermo Fisher Scientific™, USA) and NanoDropTm 2000 spectrophotometers were used for quantification and purity check of the harvested DNA, respectively.
Statistical analysis
STATA Version 14 (StataCorp LLC, TX, USA) was used for statistical analysis of data. Blood culture method was used as gold standard and compared with the developed mPCRs to assess its diagnostic performance. Sensitivity, specificity, overall agreement and area under the receiver operating characteristics (ROC) curve were calculated to establish the accuracy of the mPCR assays.
Results
Confirmation of primer specificity
Upon subjecting selected primers to the standards and known clinical strains, all 11 monoplex PCR assays could accurately detected the target pathogens, with 100% detection rate (Table 4).
Development and optimization of the mPCR assays
Optimal primer annealing temperature was achieved at 55 °C using gradient PCR. Other amplification conditions were: initial denaturation at 95 °C for 3 min; 35 cycles of denaturation at 95 °C for 30 s, annealing at 55 °C for 45 s, extension at 72 °C for 1 min, and final extension at 72 °C for 7 min. The two sets of mPCRs assays simultaneously amplified all the six and five target bacterial pathogens, respectively, using the reference strains. It also produced distinct PCR products using combined bacterial lysates, except for E. coli and Shigella spp., which shared the same target gene (uspA), Fig. 1. These results demonstrated the capability of the mPCR systems to detect all 11 targeted species, whether present in their pure forms or within mixed bacterial compositions extracted from clinical specimens. These two newly developed mPCRs, accurately amplified all 11 bacterial targeted genes in a DNA mix as well as single bacterial DNA target. This assay did not show any amplification to the five off-target bacteria (Table 1, Fig. 2).

Gels showing target genes amplified at the optimized annealing temperature (55 °C). a Multipex A: M = Marker, Lane 1 (Vibrio cholerae, (883 bP), 2. Acinetobacter baumanii (320 bp), 3. Salmonella spp. (284 bp), 4. Pseudomonas aeruginosa (190 bp), 5. Shigella spp. (128 bp), 6. E.coli(128 bp), 7. Negative control. b Multiplex B: M = Marker, 1. Enterobacter spp. (1181 bp), 2. Aeromonas spp. (891 bp), 3. Camphylobacter spp. (855 bp), 4. Providentia alcalifacience (515 bp), 5. K. pneumoniae (174 bp), 6. Negative control

Gels showing specificity of developed Mpcr. a Multipex A: l = Ladder, Lane 1. DNA mix, Lane 2 (Vibrio cholerae, 883 bP), 3. Acinetobacter baumanii (320 bp), 4. Salmonella spp. (284 bp), 5. Pseudomonas aeruginosa (190 bp), 6. E. coli (128 bp), 7–11. Off targets (Table 1) 12. Negative control. b Multiplex B: L = Ladder, Lane 1 Mixed DNA, 2. Enterobacter spp. (1181 bp), 3. Aeromonas spp. (891 bp), 4. Camphylobacter spp. (855 bp), 5. Providentia alcalifacience (515 bp), 6. K. pneumoniae (174 bp), 7–11. Off targets (Table 1) 12. Negative control
Reproducibility and LOD of the two mPCR assays for bacterial identification
The minimum detectable DNA concentration by the two mPCRs was 100 pg. The results of the reproducibility assessment yielded consistent outcomes. In each replicate run, concordant results for the presence or absence of the target bacterial strains were observed. No variations or discrepancies were detected among the replicates, suggesting good reproducibility of results fromthe mPCR assays, Fig. 3.

Gels showing the minimum detectable mixed bacterial DNA concentration. a Multiplex A mixed bacterial DNA and b multiplex B mixed bacterial DNA. a, b M (marker) lane 1. (100 ng), lane 2. (10 ng), lane 3. (1 ng), (lane 4. 100 pg), lane 5. (10 pg); lane 6. NC
Diagnostic performance of the two mPCR assays using blood samples compared to conventional blood culture
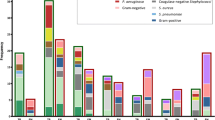
Of the 11 targeted bacterial strains, three strains were detected in patients’ blood samples by primary blood culture and mPCR, as shown in Table 5. Based on conventional culture, 15% (9/60) of the blood samples were culture-positive for mono-target bacteria, whereas 17% (10/60) were mPCR-positive. Of the ten mPCR-positive samples, two had a Salmonella spp., and Klebsiella pneumoniae co-infection. The extra detected pathogens by mPCR were 1 E. coli and 2 K. pneumonia, Table 5. The sensitivity and specificity of the mPCR using primary culture as the gold standard were 100.0% (71.7–100.0) and 98.0% (90.7–99.0), respectively (Table 5). The area under the ROC curve was 1.00 (1.00–1.00). The developed mPCR assays detected 5 pathogens (41%) out of 12 bacterial pathogens after 4 h of incubation and 12 pathogens (100%) out of 12 bacterial pathogens after 8 h of incubation (Fig. 4).

Bacterial pathogens detected in culture fluids at different incubation period
This finding highlight capacity of the mPCR assays to bridge the gap between the advantage and limitations of blood culture, to improve diagnosis of septicemia.
Discussion
This study developed two mPCR assays to detect 11 GNB pathogens commonly associated with septicemia in children. Nine identified at the genus level and two at the species level. E. coli and Shigella spp. were detected using a universal primer (uspA gene), to enhance the assays’ specificity of the assay by eliminating potential outlier annealing temperatures associated with specific E. coli and Shigella spp. genes. An identification step targeting the lacY gene present in E. coli but not in Shigellae [23] added to differentiate the organisms.
The assays’ sensitivity and specificity were 100% and 98%, respectively, with an area under ROC curve of 1.00. These assays were able to detect a co-infection that was missed by blood culture. Sensitivity was optimal due to superiority of the PCR technology, and its ability to differentiate organisms at genetic level, unlike the blood culture approach which relies on physiological characteristics, that may not necessarily be specific. The high sensitivity of the assays was further confirmed by the resulting area under ROC curve that denotes optimal accuracy and high capacity to discriminate between those with and without the targeted pathogen. The slightly sub-optimal specificity of the assays could have been due to susceptibility of the PCR method to contamination and false positives. Observed sensitivity and specificity were, however, consistent with previous studies which have found sensitivity of multiplex PCR tests for bacterial etiologies of blood stream infections among children to be higher than specificity [24]. Moreover, Cox et al. [25] observed equally high positivity and negativity upon subjecting blood culture fluid to a mPCR panel.
The high sensitivity and specificity of these assays are particularly noteworthy as it enhances its ability to detect target pathogens, enabling a definitive diagnosis. Furthermore, its capacity to detect multiple etiologies simultaneously increases its viability for application in clinical settings. Several septicemia molecular diagnostic tools, including SeptiFast, SepsiTest, SeptiCyte, U-dHRM, Prove-it, and Iridica Plex ID, have been developed to circumvent the accuracy and timeliness shortcomings of BC [26]. These kits, however, bear high purchase and operation costs, making their routine use in resource-limited countries, such as Kenya, almost impractical. Moreover, they rely on PCR-amplification and sequencing of microbial conserved genomic regions—ribosomal RNA genes and the 16S–23S inter-spacer regions; hence, turnaround time still remains high although better than that of BC [27]. Furthermore, the available multiplex PCR (mPCR) tools target one or two bacteria, which might be cumbersome, expensive and may underestimate the BSI aetiology [28].
Additional strength of the mPCR assays was its capability to detect DNA concentrations as low as 100 pg which was similar to the previous reports [29, 30], although these targeted different pathogens. This outcome is attributable to the methodology of PCR that enables it to amplify small concentrations to readily detectable quantities. The high limit of detection suggests capacity of the assays to achieve early and timely diagnosis of septicemia and their suitability as an adjunct to blood culture, especially in detection of bacterial pathogens that take long to reach detectable levels in culture.
The mPCR could not detect the target pathogens directly from whole blood samples, possibly due to low bacterial load, the small sample volume (250 µL), or potential inhibition by human DNA [31, 32]. An incubation step was, therefore, introduced before the bacterial DNA extraction to increase bacterial DNA yield. Pathogens were subsequently detectable after at least 4 h and at most 8 h of incubation. While this approach increased accuracy of the assays, it extended the overall testing process. Despite the extended process, the time-to-detection was still shorter than that of blood culture, suggesting the ability of the assays to increase utility of blood cultures to improve diagnosis especially in normally sterile body fluids.
On application of the assay to clinical testing, an overall agreement of 98.3% between blood culture and mPCR was observed. Agreement was higher than that observed in previous studies (60 to 80%) [33, 34]. The discordant case was a blood culture-negative and PCR-positive result. Since the mPCR did not miss organisms identified by blood culture, it is plausible that the mPCR assays were superior to blood culture. However, since the primary advantage of PCR-based assays is timeliness, blood culture still merits as the gold standard. Nevertheless, further larger studies can be conducted to ascertain clinical usefulness of the assays.
Only three species of pathogens, namely E. coli, Salmonella spp., and K. pneumoniae, were detected from clinical samples. This finding agrees well with previous studies that have identified these bacteria as the most common etiologies of septicemia in developing countries [35, 36]. Additionally, the mPCR assays detected co-infection in two cases involving Salmonella spp. and Klebsiella pneumoniae. Therefore, the finding of this study supports the observation that mPCR can increase diagnostic yield, by detecting co-infections that would be missed by blood culture [34].
Conclusions
The two mPCR assays demonstrated significant potential as a rapid tool for septicemia diagnosis alongside traditional blood culture method. Notably, it was able to identify additional isolates, detect co-infections, and efficiently detect low bacterial DNA loads. Validating these assays in a large and diverse study population, optimizing them to use blood samples, and incorporating GPB could improve the quality of septicemia diagnosis and patients’ management.
Study limitation
The mPCR assays could not detect pathogens directly in whole blood, which could have substantially reduced diagnosis turnaround time, and this study did not include Gram-positive bacteria that cause septicemia.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Prest J, Nguyen T, Rajah T, Prest AB, Sathananthan M, Jeganathan N. Sepsis-related mortality rates and trends based on site of infection. Crit Care Explor. 2022;4(10): e0775. https://doi.org/10.1097/CCE.0000000000000775.
Douglas NM, Hennessy JN, Currie BJ, Baird RW. Trends in bacteremia over 2 decades in the top end of the northern territory of Australia. Open Forum Infect Dis. 2020;7(11): ofaa472. https://doi.org/10.1093/ofid/ofaa472.
Ljungquist O, Blomstergren A, Merkel A, Sunnerhagen T, Holm K, Torisson G. Incidence, aetiology and temporal trend of bloodstream infections in southern Sweden from 2006 to 2019: a population-based study. Euro Surveill Bull Eur sur les Mal Transm = Eur Commun Dis Bull. 2023;28(10):2200519. https://doi.org/10.2807/1560-7917.ES.2023.28.10.2200519.
Marchello CS, Dale AP, Pisharody S, Rubach MP, Crump JA. A systematic review and meta-analysis of the prevalence of community-onset bloodstream infections among hospitalized patients in Africa and Asia. Antimicrob Agents Chemother. 2019;64(1):10–128. https://doi.org/10.1128/aac.01974-19.
Verway M, et al. Prevalence and mortality associated with bloodstream organisms: a population-wide retrospective cohort study. J Clin Microbiol. 2022;60(4): e0242921. https://doi.org/10.1128/jcm.02429-21.
WHO. Global report on the epidemiology and burden of sepsis: current evidence, identifying gaps and future directions. Geneva; 2020. https://apps.who.int/iris/bitstream/handle/10665/334216/9789240010789-eng.pdf.
Rudd KE, et al. The global burden of sepsis: barriers and potential solutions. Crit Care. 2018;22(1):232. https://doi.org/10.1186/s13054-018-2157-z.
Laupland KB, Pasquill K, Steele L, Parfitt EC. Burden of bloodstream infection in older persons: a population-based study. BMC Geriatr. 2021;21(1):31. https://doi.org/10.1186/s12877-020-01984-z.
Zasowski EJ, Claeys KC, Lagnf AM, Davis SL, Rybak MJ. Time is of the essence: the impact of delayed antibiotic therapy on patient outcomes in hospital-onset enterococcal bloodstream infections. Clin Infect Dis. 2016;62(10):1242–50. https://doi.org/10.1093/cid/ciw110.
Singh L, Das S, Bhat VB, Plakkal N. Early neurodevelopmental outcome of very low birthweight neonates with culture-positive blood stream infection: a prospective cohort study. Cureus. 2018;10(10): e3492. https://doi.org/10.7759/cureus.3492.
Tjandra KC, et al. Diagnosis of bloodstream infections: an evolution of technologies towards accurate and rapid identification and antibiotic susceptibility testing. Antibiotics. 2022;11(4):511. https://doi.org/10.3390/antibiotics11040511.
Huber S, Hetzer B, Orth-Höller D. The correct blood volume for pediatric blood cultures: a conundrum? Clin Microbiol Infect. 2020. https://doi.org/10.1016/j.cmi.2019.10.006.
Gupta E, et al. Fast track diagnostic tools for clinical management of sepsis: paradigm shift from conventional to advanced methods. Diagnostics. 2023;13(2):277. https://doi.org/10.3390/diagnostics13020277.
Klouche M, Schröder U. Rapid methods for diagnosis of bloodstream infections. Clin Chem Lab Med. 2008;46(7):888–908. https://doi.org/10.1515/CCLM.2008.157.
Afshari A, Schrenzel J, Ieven M, Harbarth S. Bench-to-bedside review: rapid molecular diagnostics for bloodstream infection—a new frontier? Crit Care. 2012;16(3):222. https://doi.org/10.1186/cc11202.
Chang S-S, et al. Multiplex PCR system for rapid detection of pathogens in patients with presumed sepsis—a systemic review and meta-analysis. PLoS ONE. 2013;8(5):1–10. https://doi.org/10.1371/journal.pone.0062323.
Dunbar SA, Gardner C, Das S. Diagnosis and management of bloodstream infections with rapid, multiplexed molecular assays. Front Cell Infect Microbiol. 2022;12: 859935. https://doi.org/10.3389/fcimb.2022.859935.
Talbert AWA, Mwaniki M, Mwarumba S, Newton CRJC, Berkley JA. Invasive bacterial infections in neonates and young infants born outside hospital admitted to a rural hospital in Kenya. Pediatr Infect Dis J. 2010;29(10):945–9.
Were T, et al. Bacteremia in Kenyan children presenting with malaria. J Clin Microbiol. 2011;49(2):671–6. https://doi.org/10.1128/JCM.01864-10.
Alemnew B, Biazin H, Demis A, Abate Reta M. Bacterial profile among patients with suspected bloodstream infections in Ethiopia: a systematic review and meta-analysis. Int J Microbiol. 2020;2020:8853053. https://doi.org/10.1155/2020/8853053.
Chaki SN, Mahande MJ. Bacteriological profile, antibiotic susceptibility pattern and predictors of bacteremia among children with bloodstream infection. Research. 2021. https://doi.org/10.21203/rs.3.rs-400910/v1.
Shen Z, et al. MPprimer: a program for reliable multiplex PCR primer design. BMC Bioinform. 2010;11(1):143. https://doi.org/10.1186/1471-2105-11-143.
Horakova K, Mlejnkova H, Mlejnek P. Specific detection of Escherichia coli isolated from water samples using polymerase chain reaction targeting four genes: cytochrome bd complex, lactose permease, beta-d-glucuronidase, and beta-d-galactosidase. J Appl Microbiol. 2008;105(4):970–6. https://doi.org/10.1111/j.1365-2672.2008.03838.x.
Straub J, et al. Diagnostic accuracy of the ROCHE Septifast PCR system for the rapid detection of blood pathogens in neonatal sepsis—a prospective clinical trial. PLoS ONE. 2017;12(11): e0187688. https://doi.org/10.1371/journal.pone.0187688.
Cox CR, Weghorn KN, Ruger K, Powers-Fletcher MV, Powell EA, Mortensen JE. Clinical utility of multiplex PCR in the detection of pathogens from sterile body fluids. J Clin Microbiol. 2024;62(4): e0161123. https://doi.org/10.1128/jcm.01611-23.
Dark P, et al. Accuracy of LightCycler®SeptiFast for the detection and identification of pathogens in the blood of patients with suspected sepsis: a systematic review and meta-analysis. Intensive Care Med. 2015;41(1):21–33. https://doi.org/10.1007/s00134-014-3553-8.
Gürtler V, Stanisich VA. New approaches to typing and identification of bacteria using the 16S–23S rDNA spacer region. Microbiology. 1996;142(1):3–16. https://doi.org/10.1099/13500872-142-1-3.
Muthumbi E, et al. Invasive salmonellosis in Kilifi, Kenya. Clin Infect Dis. 2015;61(Suppl 4):S290–301. https://doi.org/10.1093/cid/civ737.
Xu W, et al. A novel universal primer-multiplex-PCR method with sequencing gel electrophoresis analysis. PLoS ONE. 2012;7(1): e22900. https://doi.org/10.1371/journal.pone.0022900.
Li P, Zhang D, Li H, Pang J, Guo H, Qiu J. Establishment and application of multiplex PCR for simultaneously detecting Escherichia coli, Salmonella, Klebsiella pneumoniae, and Staphylococcus aureus in Minks. Front Vet Sci. 2020;7: 588173. https://doi.org/10.3389/fvets.2020.588173.
Loonen AJM, et al. Comparison of pathogen DNA isolation methods from large volumes of whole blood to improve molecular diagnosis of bloodstream infections. PLoS ONE. 2013;8(8): e72349. https://doi.org/10.1371/journal.pone.0072349.
Boardman AK, Campbell J, Wirz H, Sharon A, Sauer-Budge AF. Rapid microbial sample preparation from blood using a novel concentration device. PLoS ONE. 2015;10(2): e0116837. https://doi.org/10.1371/journal.pone.0116837.
Omar S, Murphy S, Gheevarghese R, Poppleton N. A retrospective evaluation of a multiplex polymerase chain reaction test directly applied to blood for the management of sepsis in the critically ill. S Afr J Crit Care. 2021;37(3):115–8. https://doi.org/10.7196/SAJCC.2021.v37i3.495.
Tsalik EL, et al. Multiplex PCR to diagnose bloodstream infections in patients admitted from the emergency department with sepsis. J Clin Microbiol. 2010;48(1):26–33. https://doi.org/10.1128/JCM.01447-09.
WHO. WHO sepsis technical expert meeting—meeting report. Geneva; 2018. http://apps.who.int/bookorders.
Zelellw DA, Dessie G, Worku Mengesha E, Balew Shiferaw M, Mela Merhaba M, Emishaw S. A systemic review and meta-analysis of the leading pathogens causing neonatal sepsis in developing countries. Biomed Res Int. 2021;2021:6626983. https://doi.org/10.1155/2021/6626983.
Rahn K, De Grandis SA, Clarke RC, Mcewen SA, Galin JE. Amplification of an invA gene sequence of Salmonella typhimurium by polymerase chain reaction as a specific method of detection of Salmonella. Mol Cell Probes. 1992;6:271–9.
Makki A, Wathiq G. Use of PCR to detection Pseudomonas aeruginosa from clinical samples in Hilla Teaching Hospital. J Babylon Univ Appl Sci. 2016;24(5):1414–20.
Ruwandeepika HAD, Defoirdt T, Bhowmick PP, Shekar M, Bossier P. Presence of typical and atypical virulence genes in vibrio isolates belonging to the Harveyi clade. J Appl Microbiol. 2010;109:888–99. https://doi.org/10.1111/j.1365-2672.2010.04715.x.
Datta S, Niwa H, Itoh K. Communication prevalence of 11 pathogenic genes of Campylobacter jejuni by PCR in strains isolated from humans, poultry meat and broiler and bovine faeces. J Med Microbiol. 2003;52:345–8. https://doi.org/10.1099/jmm.0.05056-0.
Rottman M. Chromosomal ampC genes in Enterobacter species other than Enterobacter cloacae, and ancestral association of the ACT-1 plasmid-encoded cephalosporinase to Enterobacter asburiae. FEMS Microbiol Lett. 2002;210:87–92.
Nhung PH, et al. Use of the novel phylogenetic marker dnaJ and DNA–DNA hybridization to clarify interrelationships within the genus Aeromonas. Int J Syst Evol Microbiol. 2007;57:1232–7. https://doi.org/10.1099/ijs.0.64957-0.
Acknowledgements
We acknowledge the Director of KEMRI for facilitating this study. We thank Kiambu County Hospital for facilitating sample collection and NUITM–KEMRI Project for providing laboratory space. We also thank Prof. Shingo Inoue for technical support.
Funding
This study was partially supported by Japan Society for Promotion of Sciences (JSPS) grant in aid for scientific research; grant number 15H05286. The funding was used for data collection and analysis.
Author information
Authors and Affiliations
Contributions
GM, EO, AM and YI—designed this study; GM developed the proposal, processed samples in the laboratory and drafted the manuscript; GM and BM analyzed data; AM, NM and YI reviewed the proposal and supervised laboratory work; AM, BM, EW, EO, DW, HS, YI inputted in the interpretation of data, development, and review of the manuscript for publication.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Approval to conduct this study was obtained from the Ethics Review Committee of Kenyatta University with the approval number (PKU/2423/11557). Parental consent was sought before recruiting patients aged below 5 years, and assent sought from the participants where applicable.
Consent for publication
All participants were informed that their information would be published, and consent to publish was granted.
Competing interests
There were no competing interests in implementation of this study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Miringu, G., Musyoki, A., Muriithi, B. et al. Development of two multiplex PCR assays for rapid detection of eleven Gram-negative bacteria in children with septicemia. Trop Med Health 52, 40 (2024). https://doi.org/10.1186/s41182-024-00606-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41182-024-00606-3