
Abstract
Traditional nuclear medicine ligands were designed to target cellular receptors or transporters with a binding pocket and a defined structure–activity relationship. More recently, tracers have been developed to target pathological protein aggregations, which have less well-defined structure–activity relationships. Aggregations of proteins such as tau, α-synuclein, and β-amyloid (Aβ) have been identified in neurodegenerative diseases, including Alzheimer’s disease (AD) and other dementias, and Parkinson’s disease (PD). Indeed, Aβ deposition is a hallmark of AD, and detection methods have evolved from coloured dyes to modern 18F-labelled positron emission tomography (PET) tracers. Such tracers are becoming increasingly established in routine clinical practice for evaluation of Aβ neuritic plaque density in the brains of adults who are being evaluated for AD and other causes of cognitive impairment. While similar in structure, there are key differences between the available compounds in terms of dosing/dosimetry, pharmacokinetics, and interpretation of visual reads. In the future, quantification of Aβ-PET may further improve its utility. Tracers are now being developed for evaluation of tau protein, which is associated with decreased cognitive function and neurodegenerative changes in AD, and is implicated in the pathogenesis of other neurodegenerative diseases. While no compound has yet been approved for tau imaging in clinical use, it is a very active area of research. Development of tau tracers comprises in-depth characterisation of existing radiotracers, clinical validation, a better understanding of uptake patterns, test-retest/dosimetry data, and neuropathological correlations with PET. Tau imaging may allow early, more accurate diagnosis, and monitoring of disease progression, in a range of conditions. Another marker for which imaging modalities are needed is α-synuclein, which has potential for conditions including PD and dementia with Lewy bodies. Efforts to develop a suitable tracer are ongoing, but are still in their infancy. In conclusion, several PET tracers for detection of pathological protein depositions are now available for clinical use, particularly PET tracers that bind to Aβ plaques. Tau-PET tracers are currently in clinical development, and α-synuclein protein deposition tracers are at early stage of research. These tracers will continue to change our understanding of complex disease processes.
Similar content being viewed by others


Introduction
Traditionally, nuclear medicine ligands were primarily designed for targeting cellular receptors or transporters. They were tightly bound, and often internalized or transported into the cell and trapped inside by metabolic transformation, while unbound ligand was cleared. More recently, a class of imaging tracers has become available whose members bind misfolded protein aggregates. This new paradigm requires different lead optimization, different types of analysis, and quantitation. Previous approaches targeted a binding pocket where derivatives of ligands displayed a defined structure-activity relationship. Examples of protein aggregate imaging include Aβ, tau, and α-synuclein. Such investigations required the design of a molecule that binds to β-sheets. The structure–activity relationship is less well defined, as no distinct binding pockets are present. Importantly, all protein depositions show a similar structural motif, and achievement of selectivity is the most important optimization goal. Nonetheless, protein sequence and aggregate structures are different enough that highly specific imaging agents have been developed for some of these targets.
Pathological protein depositions have been identified in a range of neurodegenerative diseases (Mollenhauer & Trenkwalder 2009). Tau and 43-kDa Tar DNA-binding protein (TDP-43) are present in neurofibrillary tangles characteristic of frontotemporal lobar degeneration and AD; Aβ plaques are the hallmark of AD; and α-synuclein has been identified in the pathognomonic bodies of diffuse Lewy body disease and PD. An overview of the misfolded protein depositions discussed in this article, with their associated histopathology and clinical manifestation, is presented in Fig. 1.

Schematic summary of key proteins present in frontotemporal lobar degeneration, Alzheimer’s disease, diffuse Lewy body disease and Parkinson disease. Protein monomers and their distribution for different clinical phenotypes are illustrated with symbolic drawings. Exemplary histopathology images are presented for TDP-43 inclusion, neurofibrillary tau tangles (immunohistochemistry with antibody AT8), amyloid-beta deposition (immunohistochemistry with monoclonal 6E10 Aβ antibody), and α-synuclein Lewy body inclusions (Images of TDP-43 inclusions, tau tangles, and Aβ deposits courtesy of Walter Schulz-Schaeffer, Goettingen, Germany)
Co-pathologies have also been observed, in which more than one protein forms a deposition. Identification of in vivo biomarkers for such conditions will improve diagnosis and classification of patients, provide prognostic information, and improve the efficiency of drug development. This paper will discuss some of the new positron emission tomography (PET) tracers that are being developed to target misfolded protein depositions such as Aβ, tau, and α-synuclein.
Review
Established protein tracers–detection of Aβ plaques
AD is a chronic neurodegenerative disease that can now be detected in vivo by biomarkers years before clinical manifestation. The deposition of Aβ plaques is considered one hallmark in the pathogenesis of AD, and a hypothetical model of biomarker temporal evolution has been proposed that matches the sequence of molecular events proposed in the amyloid cascade hypothesis (Jack & Holtzman 2013). The model begins with Aβ42 overproduction and aggregation, with decreased clearance, followed by plaque formation. Thus Aβ-PET and cerebrospinal fluid (CSF) Aβ42 levels are the first markers to become abnormal in AD pathogenesis, although these biomarkers are not approved for prediction of disease progression or therapeutic monitoring.
The earliest methods of detecting Aβ plaques post mortem used coloured dyes–Congo red and thioflavin T–that bind to the β-sheet structure of Aβ (Glenner 1980). Thioflavin T was used as the basis for the development of the first radiolabelled molecules for use in PET. To date, several molecules have been studied in humans (Kung 2012): 11C-Pittsburgh compound B, 18F-florbetapir, 18F-florbetaben, 18F-flutemetamol, and NAV4694. (Table 1) 18F-florbetapir, 18F-florbetaben, and 18F-flutemetamol have been approved in Europe and US for clinical use in PET evaluation of Aβ neuritic plaque density in the brains of adults who are being evaluated for AD and other causes of cognitive impairment. While these agents are becoming increasingly established in routine clinical practice, there are important learnings from their clinical development and considerations that should be taken into account for future research and development of protein deposition tracers. Some of the peculiarities are described briefly in the following paragraphs.
The three approved agents have a planar chemical structure that is suitable for binding to β-sheets in Aβ plaques. All approved agents follow the same mechanism of binding, but their different chemical structures lead to differences with regard to dosing and dosimetry; pharmacokinetics, including partitioning into grey and white matter structures; and interpretation of visual reads (Eli Lilly 2013; Piramal Imaging 2014; GE Healthcare 2014). For example, 18F-florbetapir and 18F-florbetaben PET images are approved for evaluation in greyscale, while 18F-flutemetamol PET images are read using a colour scale when used in the clinical setting. Thus each tracer requires a unique medical education programme to ensure reliable assessment of scans and to distinguish uptake in white matter from cortical grey matter.
Regulatory approval for Aβ PET scan assessment is currently based solely on a binary visual read-out, and all three reading methods have been validated against histopathology (Clark et al. 2012; Curtis et al. 2015; Sabri et al. 2015a). Of note, imaging agents used in oncology such as 18F-FDG or 18F-FLT become trapped in tumours leading to a stable or even increasing signal over time (Shields et al. 1998). In Aβ imaging, however, the tracer instead shows decreasing signal or standardised uptake values (SUVs) over time, as a result of washout after binding to Aß plaque-affected cortical areas. In addition, quantification of Aβ-PET scans typically involves calculating the SUV ratio, where the reference region is a region with a ligand uptake and washout pattern similar to Aß-plaque-affected cortical areas regardless of whether Aβ plaques are present (Schmidt et al. 2015). A number of different reference regions have been proposed (Landau et al. 2015), but further discussion is outside the scope of this review. Quantification of PET scans has the ability to better detect longitudinal changes during therapeutic intervention and has the potential for automated analysis via software with more detailed regional analysis. Future uses of Aβ-PET quantification, though not approved for routine clinical use, may include improved assessment in uncertain clinical cases, drug trial enrichment by patient selection, pre-symptomatic staging of disease, and therapeutic monitoring. Such uses require robust longitudinal assessment, reliable reference-region validation, and standardisation.
Beyond AD, amyloid-PET provides a unique opportunity for in vivo research of other conditions that are present with Aβ deposition. For example, Aβ-PET may also detect other plaque types and states of amyloid (e.g. diffuse plaques) (Sabri et al. 2015b), and thus may provide additional insights into the disease and its pathogenesis. Other conditions with Aβ-plaque depositions are reported, such as Lewy body diseases, cerebral amyloid angiopathy, brain trauma, and Down syndrome. As specific as the current tracers are for Aβ over other misfolded protein aggregates, somewhat surprisingly they do bind other amyloids outside the brain. 18F-Florbetaben and 18F-florbetapir have been reported to bind amyloid deposits in cardiac amyloidosis (Dorbala et al. 2014; Catafau & Bullich 2015; Mollee et al. 2015), and these tracers are also hypothesized to bind other peripheral amyloid deposits. In addition, tracers may also have value as a myelin biomarker in conditions such as multiple sclerosis (Matías-Guiu et al. 2015), by virtue of their white-matter signal.
Protein deposition tracers under development
Detection of tau protein
Tau protein is the name given to soluble microtubule-associated protein (MAP), which is essential for regulating intracellular transport (Spillantini & Goedert 2013). Six different isoforms of tau exist, which can be distinguished by their number of binding domains (either three or four), and different forms are accumulated in different diseases (Delacourte 1999; Braak & Braak 1998). Furthermore, hyperphosphorylation and other post-translational modifications can have an impact on tau conformation, leading to, for example, aggregation in filamentous structures.
Tau protein aggregation leads to neuronal cell dysfunction and death, and studies show a strong association between tau deposits, decreased cognitive function, and neurodegenerative changes in AD. While the evolution of AD neuropathology depends on interactions between Aβ and tau (Jucker & Walker 2011), the relative contributions of the two proteins in the development of AD remain unclear. There is emerging evidence from studying hereditary Alzheimer’s Disease (e.g. DIAN study) that continues to point to a primary role of Aß in AD. Significant proportions of the observed variance in age at symptom onset can be explained by family history and mutation type (Ryman et al. 2014). Nevertheless, several other questions remain including the presence of Aβ deposition in cognitively normal individuals and time to development of first symptoms or the weak correlation between plaque load and cognition (Morris et al. 2014). Expanding the view of the AD pathogenesis beyond Aβ and tau pathology and considering aspects such as lifestyle, cognitive reserve may provide answers in the future. Imaging Aß and tau allows investigators to look at the impact on cognition and follow subjects from an earlier stage. In addition to AD several neurodegenerative diseases – including chronic traumatic encephalopathy, progressive supranuclear palsy, corticobasal degeneration, and some variants of frontotemporal lobar degeneration – have been described in which tau aggregate deposition is a dominant pathology (Mohorko & Bresjanac 2008; Lee et al. 2001; McKee et al. 2009).
Tau is a more complex target than Aβ in that the monomer protein is much larger than Aβ, is represented in different isoforms in different diseases, is present in lower amounts and has a distinct anatomic spread throughout the brain as the different diseases progress. These characteristics, and the intracellular localisation, make the requirements for a tau PET tracer more challenging (Villemagne et al. 2015).
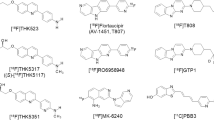
Several tau imaging compounds have been described in preclinical and clinical studies. To date, however, none have been approved. The first 18F tracer with tau binding was 18F-FDDNP, although the compound suffered from a lack of selectivity (Kepe et al. 2013). Regional uptake patterns in the brain were therefore required to differentiate Aβ and tau. Meanwhile, more-selective tracers have become available. 11C-PBB3, allows tau imaging in AD and non-AD tauopathies such as corticobasal syndrome. However, the 11C label is not preferred, as it limits widespread use due to its short half-life (20 min) (Shimada et al. 2015). Studies with the 18F-labelled tracers THK-523 and THK-5117 showed that these compounds do not correlate with Aβ distribution, but instead follow the known distribution of tau (Harada et al. 2013). However, high retention in white matter limits their use in the clinical setting. An improved compound from the series, 18F-THK-5351, provided information on tau neurofibrillary tangle pathology in living individuals in initial studies (Harada et al. 2015). The usefulness for detection of tau pathology in pure tauopathies, however, needs to be demonstrated clinically. The Siemens (now Avid) compound 18F-T808 showed good preclinical properties as well as good pharmacokinetic characteristics in a first-in-human study, although development was hampered by strong defluorination (Chien et al. 2014). Another derivative, 18F-T807 (now AV1451), showed slower kinetics but good imaging data in AD as well as in some other tauopathies. Off-target activity in the striatum and choroid plexus is, however, described for this compound (Chien et al. 2013). In comparison with other compounds, 18F-T807 has been evaluated in the most subjects. Recently presented data on three Roche tau tracers in humans showed that 18F-RO6958948 has a promising clinical profile, with good brain uptake and little retention in cognitively normal young individuals (Wong et al. 2015). The agent also has a distribution broadly consistent with published post-mortem data, including low, homogenous uptake in controls, higher, heterogeneous uptake in AD, and a different binding pattern when compared with Aβ tracers. Notably there was no apparent brain penetration of radiolabelled metabolites and no defluorination. A clinical study is ongoing to collect test-retest and whole-body dosimetry data. Furthermore, first-in-human data of the Genentech tau tracer (18F-GTP1) were recently presented, indicating a promising clinical profile (Sanabria Bohorquez et al. 2015). Finally, 18F-PI-2014 was tested recently in humans and has shown uptake in tau-target regions consistent with tau binding (Piramal Imaging, data on file). Very recently, preclinical data from 18F-MK-6240 were published (Walji et al. 2016). This 18F-labeled agent combines good in vitro characteristics for NFT binding and clean off-target profile with suitable physicochemical properties and pharmacokinetics in rhesus monkeys. A clinical study is underway and results should be expected soon. A summary of tau tracer characteristics and key features of those with published structural information is presented in Table 2.
Future development of tau tracers will require further evaluation of existing radiotracers, including preclinical characterisation, validation in the clinic, better understanding of uptake patterns in healthy controls, test-retest and human dosimetry data, and neuropathological correlations with PET, as well as head-to-head comparisons between different tracers. Improvement seems possible in the pharmacokinetic properties of 18F-labeled tracers, binding selectivity, and experience in non-AD tauopathies.
Overall, the combination of Aβ and tau-PET is currently significantly improving the knowledge of the interactions between the two proteins in humans. In addition, tau-PET–in its unique role as a marker of neurodegeneration–may allow the in vivo study of tau pathology evolution and topographic distribution across diseases. Tau imaging could also allow early, more accurate diagnosis, and more importantly monitoring of disease progression, in other tauopathies, cognitive impairment, movement disorders, and head trauma. Tau-PET may also lead to more efficient development of disease-modifying drugs not only for compounds targeting the tau protein itself.
Detection of α-synuclein
Investigation of α-synuclein and TDP-43 in post-mortem human brains has led to increased understanding of the evolution of neuropathology in PD and amyotrophic lateral sclerosis, in which lesions are believed to spread from an initial ‘seed’ of misfolded protein (Jucker & Walker 2013). There is therefore a clinical need for imaging modalities for detection of α-synuclein, which has a potential role in the differential diagnosis of PD, dementia with Lewy bodies, progressive supranuclear palsy, and multiple system atrophy. Genetic biomarkers in these conditions, while critically important in the case of inherited disease, are not salient in the majority of cases (>90 %) with sporadic PD. Detection methods for α-synuclein in CSF are currently under development, although it is not clear how CSF levels relate to histopathology data (Mollenhauer 2014) and still need further validation.
Another role for α-synuclein imaging is to decrease risk and increase efficiency in drug discovery. Imaging could identify patients early enough for potential therapies, assist with therapeutic monitoring, and enhance trial recruitment and patient enrichment. α-synuclein has advantages over dopamine as a biomarker for PD, as changes in α-synuclein may occur earlier than dopamine changes, and are not up-or down-regulated by symptomatic treatment. Efforts to develop PET or single-photon emission computed tomography tracers for α-synuclein are ongoing but are still in their infancy. The compounds currently investigated for imaging α-synuclein depositions are shown in Table 3. Research groups started with the investigation of the 18F-labeled compound BF-227 that was reported to bind to both synthetic α-synuclein aggregates as well as β-amyloid fibrils in vitro (Fodero-Tavoletti et al. 2009). It was demonstrated that BF-227 could stain α-synuclein-containing glial cytoplasmic inclusions in post-mortem tissues. Moreover, a PET study with 11C-labelled BF-227 showed its ability to detect α-synuclein deposits in the living brains of patients with multiple system atrophy (Kikuchi et al. 2010). However, the high affinity of this radiotracer for β-amyloid plaques limit its use in humans for differential diagnosis.
A series of phenothiazine derivatives was described for α-synuclein-binding (Yu et al. 2012) and the radioiodinated compound SIL23 was developed (Bagchi et al. 2013). As stated by its developers, the affinity of SIL23 for α-synuclein and its selectivity for α-synuclein versus Aβ and tau fibrils is not optimal for imaging fibrillar α-synuclein in vivo, but it could be used to screen additional ligands for suitable affinity and selectivity. Following this approach, additional compounds such as [11C] 2a and [18F] 2b have been identified that are more specific for α-synuclein and have shown the ability to cross the blood–brain barrier in animal studies (Zhang et al. 2014). However, these have not yet translated to human imaging. More recently, the same group reported the development and in vitro characterization of (benzylidene) indolin-2-one derivatives as new ligands for α-synuclein fibrils covering also PET ligands like [18F] 46a with high affinity and selectivity for α-synuclein (Chu et al. 2015). Future research will show whether some of these compounds have the ability to image α-synuclein depositions in patients.
Conclusions
Several PET tracers for detection of pathological protein depositions or aggregates are now available for routine clinical use. In particular, PET agents binding to Aβ plaques are approved as an adjunct to other diagnostic evaluations to estimate the plaque density in patients with cognitive impairment who are being evaluated for AD or other causes of cognitive decline. Tau-PET tracers are currently in clinical development, and α-synuclein protein deposition tracers are at early stage of research. Importantly, PET tracer development and imaging of protein aggregates require different approaches to those involved in imaging of receptors or transporters, including lead optimisation, scan analyses and quantitation. These tracers have, and will continue to, change our understanding of complex disease processes.
Change history
26 November 2018
The metadata in the HTML format of these original articles (Eppard et al., 2017; Decristoforo & Patt, 2017; Domnanich et al., 2017; Chan et al., 2017; Jovalekic et al., 2017; Zhang & Villalobos, 2017; Meckel et al., 2017; Li et al., 2017; Colin et al., 2017; Koziorowski et al., 2017; Ooms et al., 2017; Elsinga, 2017; Seemann et al., 2017; Müller et al., 2017; Luurtsema et al., 2017) were published with an incorrect cover date. The correct cover date is December 2016. This does not alter the text of the original articles.
Abbreviations
- Aβ:
-
Beta-amyloid
- AD:
-
Alzheimer’s disease
- CSF:
-
Cerebrospinal fluid
- PD:
-
Parkinson’s disease
- PET:
-
Positron emission tomography
- SUV:
-
Standardized uptake value
- TDP-43:
-
43 kDa Tar DNA-binding protein
- Aβ42:
-
Beta-amyloid beta (1–42)
References
Bagchi DP, Yu L, Perlmutter JS, et al. Binding of the radioligand SIL23 to a-synuclein fibrils in Parkinson’s disease tissue establishes feasibility and screening approaches for developing a Parkinson disease imaging agent. PLoS One. 2013;8:e55031.
Braak H, Braak E. Argyrophilic grain disease: frequency of occurrence in different age categories and neuropathological diagnostic criteria. J Neural Transm (Vienna). 1998;105:801–19.
Catafau AM, Bullich S. Amyloid PET imaging: applications beyond Alzheimer’s disease. Clin Transl Imaging. 2015;3:39–55.
Chien DT, Bahri S, Szardenings AK, et al. Early clinical PET imaging results with the novel PHF-tau radioligand [F-18]-T807. J Alzheimers Dis. 2013;34:457–68.
Chien DT, Szardenings AK, Bahri S, et al. Early clinical PET imaging results with the novel PHF-tau radioligand [F18]-T808. J Alzheimers Dis. 2014;38:171–84.
Chu W, Zhou D, Gaba V, et al. Design, synthesis and characterization of 3-(Benzylidene) indolin-2-one derivatives as ligands for α-synuclein fibrils. J Med Chem. 2015;58:6002–17.
Clark CM, Pontecorvo MJ, Beach TG, et al. Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid-beta plaques: a prospective cohort study. Lancet Neurol. 2012;11:669–78.
Cselényi Z, Jönhagen ME, Forsberg A, et al. Clinical validation of 18 F-AZD4694, an amyloid-β-specific PET radioligand. J Nucl Med. 2012;53(3):415–24.
Curtis C, Gamez JE, Singh U, et al. Phase 3 trial of flutemetamol labeled with radioactive fluorine 18 imaging and neuritic plaque density. JAMA Neurol. 2015;72:287–94.
Delacourte A. Biochemical and molecular characterization of neurofibrillary degeneration in frontotemporal dementias. Dement Geriatr Cogn Disord. 1999;10 Suppl 1:75–9.
Dorbala S, Vangala D, Semer J, et al. Imaging cardiac amyloidosis: a pilot study using 18F-florbetapir positron emission tomography. Eur J Nucl Med Mol Imaging. 2014;41:1652–62.
Eli L. Amyvid. Summary of product characteristics. Houten: Eli Lilly Nederland BV; 2013.
Fawaz MV, Brooks AF, Rodnick ME, et al. High affinity radiopharmaceuticals based upon lansoprazole for PET imaging of aggregated tau in Alzheimer’s disease and progressive supranuclear palsy: synthesis, preclinical evaluation, and lead selection. ACS Chem Neurosci. 2014;5:718–30.
Fodero-Tavoletti MT, Mulligan RS, Okamura N, et al. In vitro characterisation of BF227 binding to alphasynuclein/Lewy bodies. Eur J Pharmacol. 2009;617:54–8.
GE Healthcare. Vizamyl. Summary of product characteristics. Little Chalfont: GE Healthcare Limited; 2014.
Glenner GG. Amyloid deposits and amyloidosis. The beta-fibrilloses (first of two parts). N Engl J Med. 1980;302:1283–92.
Harada R, Okamura N, Furumoto S, et al. Comparison of the binding characteristics of [18 F] THK-523 and other amyloid imaging tracers to Alzheimer’s disease pathology. Eur J Nucl Med Mol Imaging. 2013;40:125–32.
Harada R, Okamura N, Furumoto S et al. 18F-THK5351: a novel PET radiotracer for imaging neurofibrillary pathology in Alzheimer's disease. J Nucl Med. 2015. Epub ahead of print.
Hashimoto H, Kawamura K, Igarashi N, et al. Radiosynthesis, photoisomerization, biodistribution, and metabolite analysis of 11C-PBB3 as a clinically useful PET probe for imaging of tau pathology. J Nucl Med. 2014;55:1532–8.
Jack Jr CR, Holtzman DM. Biomarker modeling of Alzheimer’s disease. Neuron. 2013;80:1347–58.
Jucker M, Walker LC. Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann Neurol. 2011;70:532–40.
Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. 2013;501:45–51.
Kepe V, Bordelon Y, Boxer A, et al. PET imaging of neuropathology in tauopathies: progressive supranuclear palsy. J Alzheimers Dis. 2013;36:145–53.
Kikuchi A, Takeda A, Okamura N, et al. In vivo visualization of alpha-synuclein deposition by carbon-11-labelled 2-[2-(2-dimethylaminothiazol-5-yl) ethenyl]-6-[2-(fluoro) ethoxy] benzoxazole positron emission tomography in multiple system atrophy. Brain. 2010;133(Pt 6):1772–8.
Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–19.
Kung HF. The beta-amyloid hypothesis in Alzheimer’s disease: seeing is believing. ACS Med Chem Lett. 2012;3:265–7.
Landau SM, Fero A, Baker SL, et al. Measurement of longitudinal beta-amyloid change with 18 F-florbetapir PET and standardized uptake value ratios. J Nucl Med. 2015;56:567–74.
Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–59.
Maruyama M, Shimada H, Suhara T, et al. Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron. 2013;79:1094–108.
Matías-Guiu JA, Cabrera-Martin MN, Matías-Guiu J, et al. Amyloid PET imaging in multiple sclerosis: an (18) F-florbetaben study. BMC Neurol. 2015;15:243.
McKee AC, Cantu RC, Nowinski CJ, et al. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol. 2009;68:709–35.
Mohorko N, Bresjanac M. Tau protein and human tauopathies: an overview. Zdrav Vestn. 2008;77(II):34–41.
Mollee P, Law WP, Wang WYS, Moore PT. Ng ACT. Cardiac amyloid imaging with 18F-florbetaben positron emission tomography: a pilot study. Clin Lymphoma Myeloma Leuk. 2015;15(3):e187.
Mollenhauer B. Quantification of alpha-synuclein in cerebrospinal fluid: how ideal is this biomarker for Parkinson’s disease? Parkinsonism Relat Disord. 2014;20 Suppl 1:S76–9.
Mollenhauer B, Trenkwalder C. Neurochemical biomarkers in the differential diagnosis of movement disorders. Mov Disord. 2009;24:1411–26.
Morris GP, Clark IA, Vissel B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol Commun. 2014;2:135.
Okamura N, Furumoto S, Harada R, et al. Novel 18 F-labeled arylquinoline derivatives for noninvasive imaging of tau pathology in Alzheimer disease. J Nucl Med. 2013;54:1420–7.
Okamura N, Furumoto S, Fodero-Tavoletti MT, et al. Non-invasive assessment of Alzheimer’s disease neurofibrillary pathology using 18 F-THK5105 PET. Brain. 2014;137:1762–71.
Okamura N, Furumoto S, Harada R, et al. In vivo selective imaging of tau pathology in Alzheimer’s disease with 18F-THK5117. J Nucl Med. 2015;55(1):136.
Piramal Imaging. Neuraceq. Summary of product characteristics. Havant: Piramal Imaging Limited; 2014.
Ryman DC, Acosta-Baena N, Aisen PS, et al. Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta-analysis. Neurology. 2014;83(3):253–60.
Sabri O, Sabbagh MN, Seibyl J, et al. Florbetaben PET imaging to detect amyloid plaques in Alzheimer disease: Phase 3 study. Alzheimers Dement. 2015a;11:964–74.
Sabri O, Catafau A, Barthel H, et al. Impact of morphologically distinct amyloid β (Aβ) deposits on 18F-florbetaben (FBB) PET scans. J Nucl Med. 2015b;56(3):195.
Sanabria Bohorquez S, Barret O, Tamagnan G, et al. Identification and first-in-human evaluation of Genentech Tau Probe 1 ([18F] GTP1). Barcelona, Spain: Poster presented at the 8th Clinical Trials on Alzheimer’s Disease; 2015. Abstract P2-21.
Schmidt ME, Chiao P, Klein G, et al. The influence of biological and technical factors on quantitative analysis of amyloid PET: Points to consider and recommendations for controlling variability in longitudinal data. Alzheimers Dement. 2015;11:1050–68.
Shao X, Carpenter GM, Desmond TJ, et al. Evaluation of [(11) C] N-methyl lansoprazole as a radiopharmaceutical for PET imaging of tau neurofibrillary tangles. ACS Med Chem Lett. 2012;3:936–41.
Shields AF, Grierson JR, Dohmen BM, et al. Imaging proliferation in vivo with [F-18] FLT and positron emission tomography. Nat Med. 1998;4:1334–6.
Shimada H, Shinotoh H, Sahara N, et al. Diagnostic utility and clinical significance of tau PET imaging with [11C] PBB3 in diverse tauopathies. Miami, FL, USA: Presented at the 9th Human Amyloid Imaging Conference; 2015.
Shoghi-Jadid K, Small GW, Agdeppa ED, et al. Localization of neurofibrillary tangles and beta-amyloid plaques in the brains of living patients with Alzheimer disease. Am J Geriatr Psychiatry. 2002;10:24–35.
Small GW, Kepe V, Siddarth P, et al. PET scanning of brain tau in retired national football league players: preliminary findings. Am J Geriatr Psychiatry. 2013;21:138–44.
Smid LM, Kepe V, Vinters HV, et al. Postmortem 3-D brain hemisphere cortical tau and amyloid-beta pathology mapping and quantification as a validation method of neuropathology imaging. J Alzheimers Dis. 2013;36:261–74.
Spillantini MG, Goedert M. Tau pathology and neurodegeneration. Lancet Neurol. 2013;12:609–22.
Thompson PW, Ye L, Morgenstern JL, et al. Interaction of the amyloid imaging tracer FDDNP with hallmark Alzheimer’s disease pathologies. J Neurochem. 2009;109:623–30.
Villemagne VL, Fodero-Tavoletti MT, Masters CL, Rowe CC. Tau imaging: early progress and future directions. Lancet Neurol. 2015;14:114–24.
Walji AM, Hostetler ED, Selnick H et al. Discovery of 6-(fluoro-18F)-3-(1H-pyrrolo [2,3-c] pyridin-1-yl) isoquinolin-5-amine ([18F]-MK-6240): A Positron Emission Tomography (PET) Imaging Agent for Quantification of Neurofibrillary Tangles (NFTs) J Med Chem. 2016 Apr 18. [Epub ahead of print]
Wong DF, Borroni D, Kuwabara H, et al. First in-human PET study of 3 novel Tau radiopharmaceuticals: [11C] RO6924963, [11C] RO6931643, and [18 F] RO6958948. Washington, DC, USA: Presented at AAIC; 2015.
Xia CF, Arteaga J, Chen G, et al. [(18) F] T807, a novel tau positron emission tomography imaging agent for Alzheimer’s disease. Alzheimers Dement. 2013;9:666–76.
Yu L, Cui J, Padakanti PK, et al. Synthesis and in vitro evaluation of alpha-synuclein ligands. Bioorg Med Chem. 2012;20:4625–34.
Zhang X, Jin H, Padakanti PK, et al. Radiosynthesis and Evaluation of Two PET Radioligands for Imaging alpha-Synuclein. Appl Sci (Basel). 2014;4:66–78.
Acknowledgements
Medical writing assistance was provided by Dan Booth PhD (Bioscript Medical Ltd) and funded by Piramal Imaging GmbH, Berlin, Germany. Histopathology images of TDP-43 inclusions, Tau tangles and Aβ deposits were kindly provided by Professor Walter Schulz-Schaeffer (Goettingen, Germany).
Authors’ contributions
AJ, NK, AM, and AS developed the presentation on which this manuscript is based, and were involved in the drafting and final approval of the manuscript. All authors read and approved the final manuscript.
Competing interests
AJ is an employee of Piramal Imaging GmbH, Berlin. NK, AM, and AS are employees of, and holds property rights/patents for (radio) pharmaceuticals with, Piramal Imaging GmbH, Berlin.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Jovalekic, A., Koglin, N., Mueller, A. et al. New protein deposition tracers in the pipeline. EJNMMI radiopharm. chem. 1, 11 (2017). https://doi.org/10.1186/s41181-016-0015-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41181-016-0015-3