
Abstract
Background
miRNAs are a type of conserved, small RNA molecule that regulate gene expression and play an important role in the growth and development of plants. miRNAs are involved in seed germination, root development, shoot apical meristem maintenance, leaf development, and flower development by regulating various target genes. However, the role of miRNAs in the mechanism of tea plant flower sterility remains unclear. Therefore, we performed miRNA sequencing on the flowers of fertile male parents, female parents, and sterile offspring.
Results
A total of 55 known miRNAs and 90 unknown miRNAs were identified. In the infertile progeny, 37 miRNAs were differentially expressed; 18 were up-regulated and 19 were down-regulated. miR156, miR157, miR164, miR167, miR169, miR2111 and miR396 family members were down-regulated, and miR160, miR172 and miR319 family members were up-regulated. Moreover, we predicted that the 37 differentially expressed miRNAs target a total of 363 genes, which were enriched in 31 biological functions. We predicted that miR156 targets 142 genes, including ATD1A, SPL, ACA1, ACA2, CKB22 and MADS2.
Conclusion
We detected a large number of differentially expressed miRNAs in the sterile tea plant flowers, and their target genes were involved in complex biological processes. Among these miRNAs, the down-regulation of miR156 may be one of the factor in the formation of sterile floral buds in tea plants.
Similar content being viewed by others


Introduction
Tea is one of the most popular and widely consumed beverages in the world. As such, producers expect tea varieties to have high yield [1]. However, the process of tea plant flowering indicates that the energy of nutrition turns to reproductive growth, which will compete for nutrition and lead to a reduction in production. Therefore, investigating the mechanism of tea plant sterility may play a key role in increasing the yield of tea. Among the factors that cause flower sterility, the abortion or degradation of stamens and pistils is an important aspect that causes plants to produce sterile flowers, and its molecular mechanisms have attracted much attention [2].
MicroRNAs (miRNAs) are a type of endogenous, non-coding, single-stranded RNA molecule composed of about 21 to 25 nucleotides (nt) in length that exist widely across organisms and are quite conserved [3]. Generally, miRNAs recognize their target gene mRNAs through complementary pairing with the mRNA, and degrade or inhibit the translation of the mRNA, thereby regulating the abundance and function of the target mRNA [4]. A miRNA can usually regulate multiple target genes, and the same target gene may also be regulated by multiple miRNAs [5]. Researchers have discovered that miRNAs are involved in regulating plant biological processes, such as leaf development [6], anther development [7], cell differentiation [8], flowering [9], floral organ morphology development [10], and response to environmental stress [11]. In addition, mutations in miRNA target genes cause defects in plant development [12], indicating that miRNAs play an important role in promoting plant survival.
Studies have shown that a number of miRNAs, including miR156, miR159, miR160, miR164, Mir166/165, miR167, miR169, miR172, miR319, miR390, and miR399, are related to flower development. It has been reported that miR156, miR172 and miR390 are involved in the regulation of flowering time; miR156 was up-regulated at the juvenile stage to prevent flowering, followed by a decrease in expression with plant growth [13]; miR172 acts downstream of miR156, SPL9 and SPL10, and was regulated by miR156, contrary to the expression pattern of miR156 [14]; mutations in miR164 affected the development of the carpel [15]; miR167 can regulate the expression of ARF6 and ARF8 in specific floral organs, but different isoforms showed differences in the ability to inhibit ARF6 and ARF8 expression [16]. Similar to miR159, up-regulation of miR319 causes stamen defects, leading to sterility [17]. Moreover, auxin response elements have been shown to up-regulate the expression of miR160, while miR160 down-regulated the expression of the genes ARF10, ARF16 and ARF17 [18].
In our study, we selected Foxiang2 (FBH), Fudingbaicha (MBH), and their hybrid sterile progeny (ZDH) because of the self-incompatibility of tea plants. We performed small RNA sequencing on flowers, analyzed the differentially expressed genes, and predicted the target genes of different miRNAs. Through the analysis of the data, we identified the key miRNAs related to tea plant sterility and provided reference information for research on the mechanism of tea plant sterility .
Materials and methods
Plant materials
The tea samples (C. sinensis (L.) O. Kuntze) used in this experiment were ‘Foxiang2’ (FBH), ‘Fudingbaicha’ (MBH) and sterile hybrid (ZDH), were collected from the Tea Research Institute of Yunnan Academy of Agricultural Sciences. The floral organ of ‘Foxiang2’ and ‘Fudingbaicha’ were complete bisexual flowers. The hybrid offspring of sterile flower buds consist of multiple dwarf pistils, the petals cannot expand normally, the filaments were short, and no pollen in the anthers (Fig. 1). The buds of the samples were collected during the flowering period, and three biological replicates were obtained in each group. The samples were frozen in liquid nitrogen and refrigerated at − 80 °C.

Morphological characteristics of the tea flower
RNA library construction and sequencing
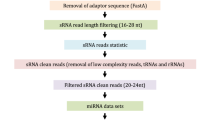
Small RNA library construction and sequencing were conducted by Novogene Technology (Beijing, China). The Small RNA Sample Pre-Kit was used to construct the library. Briefly, with total RNA as the starting material, the special structure of the 3′ and 5′ ends of Small RNA were used to add linkers to both ends of the Small RNA, followed by reverse transcription to synthesize cDNA to construct the library. The effective library concentration was greater than 2 nM. Agilent 2100 was used to detect the insert size of the library and Illumina HiSeq 2000 sequencing was performed. All of the experiments were performed in triplicate. In order to ensure the quality of information analysis, the following was removed: low-quality reads, reads with less than 10% base information, reads contaminated with 5′ linkers, reads without 3′ linker sequences and inserts, 3′ linker sequences, and polyA/T/G/C reads.
miRNA analysis
The Bowtie software package locates the small RNA after length screening to the reference sequence, and analyzes the distribution of small RNAs on the reference sequence [19]. Then, the reads mapped to the reference sequence are compared with the specified range sequence in miRBase to obtain the details of the matched small RNA of each sample, including the secondary structure of the known miRNA on the match, the sequence of the miRNA in each sample, and information, such as length and number of occurrences. The signature hairpin structure of miRNA precursors was used to predict new miRNAs. The comparison and annotation of all small RNAs and various RNAs were then summarized. Total rRNA in the classification annotation results can be used as a quality control standard for a sample, and the proportion of the total rRNA in plant samples should be less than 60%.
Prediction and analysis of target genes
To obtain the corresponding relationship between miRNA and target gene, the target genes of known and novel miRNAs were predicted using the miRNA target gene prediction software psRobot [20]. Gene Ontology (GO) enrichment analysis was performed on each set of differentially expressed miRNA target genes. GO enrichment analysis was based on Wallenius non-centralhyper-geometric distribution.
Analysis of differentially expressed miRNAs
We performed statistical analysis on the expression of known and new miRNAs in each sample. The input data of differential expression is the read count data obtained from miRNA expression level analysis. For the biological replicates, DESeq2 based on negative binomial distribution was used for analysis. In the comparison of the two samples, the binomial distribution was emphasized, and A1 and A2 were the total number of reads. Define M = (log2A1-log2A2) and A = (log2A1 + log2A2/2). DESeq2 calculates the difference expression based on the MA-plot [21]. The resulting P-values were adjusted using the Benjamin and Hochberg approach for controlling the false discovery rate. The genes with P-value < 0.05 found by DESeq were differentially expressed [22].
Quantitative real-time PCR assays
Total RNA was isolated using TRlpure reagent (BioTeke, China) according to the manufacturer’s instructions. cDNA was synthesized from total RNA using a PrimeScript RT reagent kit (TaKaRa, Japan). The obtained cDNA was used as a template in a SYBR green-basedq-PCR reaction kit (CFX-96, Bio-Rad, Hercules, CA, USA). miR165a-3p was used for normalization, and the primers are shown in Table 1.
Results
Small RNA sequencing data
In order to explore the role of miRNAs in the regulation of tea plant flower development, we used high-throughput sequencing to construct miRNA libraries of FBH, MBH and ZDH. The Q20 percentage of raw data exceeded 98%, and the Q30 percentage was above 96%, with the ratios of GC content above 47%. As a result, a total of 13,976,247, 16,644,199, and 13,976,692 clean reads were obtained for FBH, MBH and ZDH, respectively. The length of plant small RNA ranged from 18 ~ 30 nt, and a total of 11,704,253, 14,693,758 and 13,045,116 small RNA sequence read were identified for FBH, MBH and ZDH, respectively (Table 2). The results showed that the small RNA in the three miRNA libraries were mainly distributed in the range of 21 ~ 24 nt, of which 24 nt was the most abundant (Fig. 2). In regard to the position of the small RNA after length screening to the reference sequence, the ratio of mapped small RNA for for FBH, MBH and ZDH was 60.49, 53.61 and 56.4%, respectively. The number of known miRNAs were 108,703, 134,730 and 105,547, novel miRNA were 64,681, 74,929 and 53,515, rRNA were 411,500, 420,492 and 295,026, ta-siRNA were 33,333, 40,746 and 21,517, snRNA were 8441, 8086 and 2975, and snoRNA were 4114, 3020 and 3227, resoectively (Table 3).

Distribution of small RNA length. The abscissa represents the number of bases, and the ordinate represents the number of redundant reads
Screening and analysis of the miRNAs
To identify known miRNAs, we compared our data with known miRNA data of Arabidopsis in the miRBase 21.0 database (http://www.mirbase.org/ftp.shtml). A total of 55 known miRNAs were identified and divided into 27 miRNA families. Among these miRNA families, miR156 and miR396 contained four members, miR159, miR166, miR169, miR172 and miR399 contained three members, miR157, miR160, miR164, miR167, miR169, miR170, miR171, miR319, miR390, miR395, miR398 and miR858 contained 2 members, and miR162, miR165, miRK2111, miR393, miR394, miR403, miR408 and miR8175 contained 1 member (Table S1). In addition, we identified 90 putative novel miRNAs (Table S2). All novel miRNA sequences were 19 to 25 nt in length, of which the largest proportion, of miRNAs, accounting for 40.00%, were 24 nt. Interestingly, the sequence with A as the first base accounted for 44% of the novel miRNAs, and the sequence with U as the first base comprised the largest proportion of known miRNAs (Fig. 3).

The number of bases in the first position of the miRNA sequence. Among the known miRNAs, there were 37 miRNAs whose first base was U, 7 with C, 7 with G, and 4 with A. Among the novel miRNAs, there were 40 miRNAs whose first base was A, 34 with U, 9 with C, and 7 with G
Differentially expressed miRNAs
The expression level of the known and new miRNA in each sample was measured, and the expression level was normalized with TPM [23]. A total of 104 differentially expressed miRNAs were detected in FBH, MBH and ZDH (Fig. 4A). Venn diagram analysis revealed the unique and shared miRNAs in the samples (Fig. 4B). There were 86 differentially expressed miRNAs in common between FBH and ZDH, and 66 differentially expressed miRNAs in common between MBH and ZDH. Notably, 37 differentially expressed miRNAs identified in ZDH, and their expression was significantly different from FBH and MBH, but there was no difference between FBH and MBH. Among these 37 miRNAs, 18 miRNAs were up-regulated and 19 miRNAs were down-regulated. Twelve of the miRNAs that were down-regulated included miR156, miR157, miR164, miR167, miR169, miR2111 and miR396 family members, and 6 of the up-regulated miRNAs included miR160, miR172 and miR319 family members (Fig. 4C) (Table 4).

Analysis of differentially expressed miRNAs. A miRNA cluster analysis, clustering by log10(TPM + 1) value, where red indicates high expression miRNA and blue indicates low expression miRNA. B Venn diagram showing the number of differentially expressed miRNA. C Relative expression heat map of known miRNAs, where pink represents high expression and green represents low expression
Analysis of miRNA target genes
In order to investigate the regulatory effect of the miRNAs on gene expression, the psRobot software was used to predict the target genes by analyzing the known and novel miRNAs. A total of 145 miRNAs were analyzed and 4007 genes were predicted. Among the predicted genes, 1200 genes were annotated, including WDR, MYB, bHLH, SPL, WRKY, NAC, APETALA2, AGL, ARF, AP2, as well other transcription factors. The results indicated that miRNAs target multiple genes, and one gene is targeted by multiple miRNAs, suggesting that miRNAs can regulate multiple functions, and that miRNAs have the same or different regulatory effects in tea plants.
In addition, 37 differentially expressed miRNAs have 363 annotated target genes. In order to study the function of miRNAs further, the characteristics of the 363 target genes were analyzed through the GO database, including three biological processes, cellular components, and molecular function. The results showed that a total of 31 subcategories were enriched, and “transcription” and “regulation of transcription” were the most enriched biological process. Cellular component analysis showed that “nucleus”, “cytoplasm” and “plasma membrane” were the most representative subcategories. “DNA binding”, “metal ion binding”, protein binding” and “transcription factor activity” were the most representative groups of molecular function. Furthermore, 3, 3, 2, and 4 genes were enriched in “photoperiodism/flowering”, “negative regulation of flower development”, “calcium ion transmembrane transport”, and “calmodulin binding”, respectively (Fig. 5).

GO enrichment analysis of the differentially expressed miRNA. Orange indicates biological processes, blue indicates cellular components, and gray indicates molecular functions
Among the target genes, three auxin response factor genes (ARF18) targeted by miR160a-5p were predicted; nine floral homeotic protein (AP2) and two ethylene-responsive transcription factor (RAP2–7) co-targeted by miR172a, miR172c and miR172e-3p were predicted; two transcription factors (GAMYB) targeted by miR319c were predicted; two calcium-transporting ATPases (ACA12) co-targeted by miR396a-5p and miR396b-5p were predicted; and one ABC transporter I family member (ABCI11) co-targeted by miR172a and miR172c were predicted. In addition, we predicted that miR156 has 142 target genes, including three ATPase family AAA domain-containing proteins (ATD1A), four Squamosa promoter-binding proteins (SPL), two calcium-transporting ATPases (ACA1 and ACA2), two Cyclin-dependent kinases (CKB22), and Floral homeotic protein (MADS2). These results indicated that the differentially expressed miRNAs regulate complex biological processes.
qPCR analysis of miRNA
We randomly selected 12 miRNAs for RT-qPCR analysis to verify the accuracy of the small RNA sequencing. Among these miRNAs, miR156a-5p, miR156j, miR157a-5p, miR157d, miR164a and miR164c-5p were down-regulated, and miR319, amiR319, cmiR172a, miR172e-3p, miR160a-5p and novel_126 were up-regulated in ZDH (Fig. 6). The expression level of these miRNAs in FBH, MBH and ZDH was consistent with the change trend of small RNA sequencing analysis.

qPCR analysis of selected differentially expressed miRNAs. Data is presented as the mean ± SD, and n = 3 independent experiments
Discussion
As the reproductive structure of plants, flowers are the key to species reproduction. Flower development is regulated by external factors, such as the environment, and the plant’s genetic information [24]. MicroRNAs play an important role in gene regulation and participate in various biological functions by degrading target gene transcripts or disturbing translation [25]. Here, we selected FBH, MBH and ZDH for small RNA sequencing analysis. By narrowing the scope of differentially expressed miRNAs, the miRNAs related to infertility can be accurately discovered. A total of 55 known miRNAs and 90 novel miRNAs were identified, and 4007 target genes were predicted. Additionally, 37 differentially expressed miRNAs were screened and 363 miRNA target genes were predicted. Among them, miR156, miR160, miR164, miR167, miR169, miR172 and miR319 families have been confirmed to be involved in regulating the formation and development of floral organs.
In Arabidopsis, miR160 participates in the auxin-related signal transduction pathway through targeted regulation of the ARF10, ARF16 and ARF17 genes, and regulates the growth and development of flowers [26]. miR172 controls sex differentiation and meristem cells by targeting AP2-like [27, 28]. Overexpression of miR172 can reduce the level of AP2 protein and also produces abnormal flower phenotypes [29]. The absence of miR319a cause defects in the development of petals and stamens, such as abnormal petals and impaired anther formation [30]. In addition, miR319 regulates the morphology of petals and stamens by regulating the TCP family [31]. Overexpression of the TCP24 gene in Arabidopsis destroys the cell wall of anther endoderm hierarchy, leading to male sterility [32]. In contrast, the expression of miR160, miR172 and miR319 in ZDH was significantly up-regulated. As such, we concluded that the up-regulation of miR160, miR172 and miR319 may cause tea plant sterility.
miR156 inhibits the expression of the SPL transcription factor family to regulate the formation and development of floral organs [33], and 11 genes in the SPL family are regulated by miR156. A previous study reported that miR156 controls the elongation of Arabidopsis petals and sepals by regulating the expression of the SPL2 gene [34]. SPL8 gene deletion in Arabidopsis results in small anthers, less pollen, and reduced fertility [35]. Notably, miR172 influences floral organ identity by regulating AP2 [36], and miR156 positively regulates the expression of miR172 [37]. Our results revealed that miR156 and miR172 had opposite expression patterns in ZDH compared with FBH and MBH, suggesting that miR156 and miR172 are involved in another regulatory mechanism in producing sterile tea plant flowers. In addition, miR164 targets NAC transcription factors, including CUC1 and CUC2, which can promote the development of floral organs [38]. In double mutants of CUC2 and CUC1, floral organ fusion occurred, and the number of pistil and stamen were reduced [39]. We identified that the expression of miR156 and miR164 families were down-regulated in sterile flowers, indicating that miR156 and miR164 also play a key role in the mechanism of sterility.
miR159 and miR319 have overlapping effects in the regulation of flower development, regulating flowering time and anther development [17]. In addition, miR2118 also regulates the morphology and development of anthers [40]. In our results, the miR159 and miR2118 families, which have been reported to be related to flower development, were not differentially expressed. This indicates that they were not involved in the formation of sterile floral buds in this study.
In conclusion, Fig. 7 showed a schematic representation of the proposed molecular basis of the above results and published literature [41] on sterile floral buds. According to previous studies, we found that the SPL family of genes were up-regulated in ZDH [41]. Combined with the results, We hypothesize that the down-regulation of miR156 and the decreased inhibitory effect lead to overexpression of SPL. In contrast to the previous reports, up-regulation of miR172 did not inhibit AP2 expression. In addition, miR160, miR164, and miR319 were also involved in regulating the formation of sterile floral buds. Our study identified a number of important miRNAs related to tea plant fertility, these results provided a basis for further elucidating the mechanism of sterile floral buds in tea plant.

Schematic representation of the pathway for sterile floral buds in tea plants
Availability of data and materials
We have provided detailed information about the materials and methods in our manuscript.
Abbreviations
- FBH:
-
Foxiang2
- MBH:
-
Fudingbaicha
- ZDH:
-
Hybrid sterile flowers
- GO:
-
Gene ontology
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- Q20:
-
Quality score 20
- TPM:
-
Transcripts per million clean tags
References
Wei C, Yang H, Wang S, Zhao J, Liu C, Gao L, et al. Draft genome sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality. Proc Natl Acad Sci U S A. 2018;115(18):e4151–8. https://doi.org/10.1073/pnas.1719622115.
Smith AR, Zhao D. Sterility caused by floral organ degeneration and abiotic stresses in Arabidopsis and cereal grains. Front Plant Sci. 2016;7:1503.
Sun F, Guo G, Du J, et al. Whole-genome discovery of miRNAs and their targets in wheat (Triticum aestivum L.). BMC Plant Biol. 2014;14(1):142. https://doi.org/10.1186/1471-2229-14-142.
Baulcombe D. RNA silencing in plants. Nature. 2004;431(7006):356–63. https://doi.org/10.1038/nature02874.
Yu J, Zhang Z, Huang S, et al. Analysis of miRNAs targeted storage regulatory genes during soybean seed development based on transcriptome sequencing. Genes. 2019;10(6):408. https://doi.org/10.3390/genes10060408.
Mecchia MA, Debernardi JM, Rodriguez RE, Schommer C, Palatnik JF. MicroRNA miR396 and RDR6 synergistically regulate leaf development. Mech Dev. 2013;130(1):2–13. https://doi.org/10.1016/j.mod.2012.07.005.
Yang X, Zhao Y, Xie D, Sun Y, Zhu X, Esmaeili N, et al. Identification and functional analysis of microRNAs involved in the anther development in cotton genic male sterile line Yu98-8A. Int J Mol Sci. 2016;17(10):1677. https://doi.org/10.3390/ijms17101677.
Rodriguez RE, Schommer C, Palatnik JF. Control of cell proliferation by microRNAs in plants. Curr Opin Plant Biol. 2016;34:68–76. https://doi.org/10.1016/j.pbi.2016.10.003.
Huo H, Wei S, Bradford KJ. Delay of germination1 (DOG1) regulates both seed dormancy and flowering time through microRNA pathways. Proc Natl Acad Sci. 2016;113(15):e2199–206. https://doi.org/10.1073/pnas.1600558113.
Nag A, Jack T. Sculpting the flower; the role of microRNAs in flower development. Curr Top Dev Biol. 2010;91:349–78. https://doi.org/10.1016/S0070-2153(10)91012-0.
Ohama N, Sato H, Shinozaki K, Yamaguchi-Shinozaki K. Transcriptional regulatory network of plant heat stress response. Trends Plant Sci. 2017;22(1):53–65. https://doi.org/10.1016/j.tplants.2016.08.015.
Singh A, Gautam V, Singh S, Sarkar Das S, Verma S, Mishra V, et al. Plant small RNAs: advancement in the understanding of biogenesis and role in plant development. Planta. 2018;248(3):545–58. https://doi.org/10.1007/s00425-018-2927-5.
Huijser P, Schmid P. The control of developmental phase transitions in plants. Development. 2011;138(19):4117–29. https://doi.org/10.1242/dev.063511.
Wu G, Park Y, Conway SR, et al. The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell. 2009;138(4):750–9. https://doi.org/10.1016/j.cell.2009.06.031.
Baker CC, Sieber P, Wellmer F, Meyerowitz EM. The early extra petals1 mutant uncovers a role for microRNA miR164c in regulating petal number in Arabidopsis. Curr Biol. 2005;15(4):303–15. https://doi.org/10.1016/j.cub.2005.02.017.
Wu G, Poethig RS. Temporal regulation of shoot development in Arabidopsis thaliana by miR156 and its target SPL3. Development. 2006;133(18):3539–47. https://doi.org/10.1242/dev.02521.
Palatnik JF, Wollmann H, Schommer C, Schwab R, Boisbouvier J, Rodriguez R, et al. Sequence and expression differences underlie functional specialization of Arabidopsis microRNAs miR159 and miR319. Dev Cell. 2007;13(1):115–25. https://doi.org/10.1016/j.devcel.2007.04.012.
Liu X, Huang J, Wang Y, Khanna K, Xie Z, Owen HA, et al. The role of floral organs in carpels, an Arabidopsis loss-of-function mutation in MicroRNA160a, in organogenesis and the mechanism regulating its expression. Plant J. 2010;62(3):416–28. https://doi.org/10.1111/j.1365-313X.2010.04164.x.
Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. https://doi.org/10.1186/gb-2009-10-3-r25.
Wu HJ, Ma YK, Chen T, et al. PsRobot: a web-based plant small RNA meta-analysis toolbox. Nucleic Acids Res. 2012; Jul;40:W22–8.
Zeng W, Sun Z, Lai Z, Yang S, Chen H, Yang X, et al. Determination of the MiRNAs related to bean Pyralid larvae resistance in soybean using small RNA and transcriptome sequencing. Int J Mol Sci. 2019;20(12):2966. https://doi.org/10.3390/ijms20122966.
Storey JD. The positive false discovery rate: a Bayesian interpretation and the q-value. Ann Stat. 2003;31:2013–35.
Zhou L, Chen JH, Li ZZ, et al. Integrated profiling of microRNAs and mRNAs: microRNAs located on Xq27.3 associate with clear cell renal cell carcinoma. PLoS One. 2010;5:e15224.
Hong Y, Jackson S. Floral induction and flower formation--the role and potential applications of miRNAs. Plant Biotechnol J. 2015;13(3):282–92. https://doi.org/10.1111/pbi.12340.
Spanudakis E, Jackson S. The role of microRNAs in the control of flowering time. J Exp Bot. 2014;65(2):365–80. https://doi.org/10.1093/jxb/ert453.
Qiao M, Zhao ZJ, Song YG, Liu Z, Cao L, Yu Y, et al. Proper regeneration from in vitro cultured Arabidopsis thaliana requires the microRNA-directed action of an auxin response factor. Plant J. 2012;71(1):14–22. https://doi.org/10.1111/j.1365-313X.2012.04944.x.
Chuck G, Meeley R, Hake S. Floral meristem initiation and meristem cell fate are regulated by the maize AP2 genes ids1 and sid1. Development. 2008;135(18):3013–9. https://doi.org/10.1242/dev.024273.
Sieber P, Wellmer F, Gheyselinck J, Riechmann J́L, Meyerowitz EM. Redundancy and specialization among plant microRNAs: role of the MIR164 family in developmental robustness. Development. 2007;134(6):1051–60. https://doi.org/10.1242/dev.02817.
Aukerman MJ, Sakai H. Regulation of flowering time and floral organ identity by a microRNA and its APETALA2-like targetgenes. Plant Cell. 2003;15(11):2730–41. https://doi.org/10.1105/tpc.016238.
Nag A, King S, Jack T. miR319a targeting of TCP4 is critical for petal growth and development in Arabidopsis. Proc Natl Acad Sci U S A. 2009;106(52):22534–9. https://doi.org/10.1073/pnas.0908718106.
Li ZY, Li B, Shen WH, Huang H, Dong A. TCP transcription factors interact with AS2 in the repression of class-I KNOX genes in Arabidopsis thaliana. Plant J. 2012;71(1):99–107. https://doi.org/10.1111/j.1365-313X.2012.04973.x.
Wang H, Mao YF, Yang J, et al. TCP24 modulates secondary cell wall thickening and anther endothecium development. Front Plant Sci. 2015;6:436.
Chen GS, Li JT, Liu Y, et al. SPLRoles of the GA-mediated gene family and miR156 in the floral development of Chinese chestnut. Int J Mol Sci. 2019;20(7):1577. https://doi.org/10.3390/ijms20071577.
Wang ZS, Wang Y, Kohalmi SE, et al. SQUAMOSA PROMOTER BINDING PROTEIN-LIKE 2 controls floral organ development and plant fertility by activating ASYMMETRIC LEAVES 2 in Arabidopsis thaliana. Plant Mol Biol. 2016;92(6):661–74. https://doi.org/10.1007/s11103-016-0536-x.
Zhang Y, Schwarz S, Saedler H, Huijser P. SPL8, a local regulator in a subset of gibberellin-mediated developmental processes in Arabidopsis. Plant Mol Biol. 2007;63(3):429–39. https://doi.org/10.1007/s11103-006-9099-6.
Gandikota M, Birkenbihl RP, Höhmann S, Cardon GH, Saedler H, Huijser P. The miRNA156/157 recognition element in the 3′ UTR of the Arabidopsis SBP box gene SPL3 prevents early flowering by translational inhibition in seedlings. Plant J. 2007;49(4):683–93. https://doi.org/10.1111/j.1365-313X.2006.02983.x.
Wu G, Park MY, Conway SR, Wang JW, Weigel D, Poethig RS. The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell. 2009;138(4):750–9. https://doi.org/10.1016/j.cell.2009.06.031.
González-Carranza ZH, Zhang X, Peters JL, Boltz V, Szecsi J, Bendahmane M, et al. HAWAIIAN SKIRT controls size and floral organ number by modulating CUC1 and CUC2 expression. PLoS One. 2017;12(9):e0185106. https://doi.org/10.1371/journal.pone.0185106.
Cucinotta M, Manrique S, Cuesta C, Benkova E, Novak O, Colombo L. CUP-SHAPED COTYLEDON1 (CUC1) and CUC2 regulate cytokinin homeostasis to determine ovule number in Arabidopsis. J Exp Bot. 2018;69(21):5169–76. https://doi.org/10.1093/jxb/ery281.
Araki S, Le NT, Koizumi K, et al. miR2118-dependent U-rich phasiRNA production in rice anther wall development. Nat Commun. 2020;11:3115.
Chen L, Qu H, Xia L, et al. Transcriptome profiling of the fertile parent and sterile hybrid in tea plant flower buds. Hereditas. 2019;156:12.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (32060699, U20A2045).
Funding
This work was funded by NSFC32060699.
Author information
Authors and Affiliations
Contributions
LC conceived and designed the experiments. YL, HJ and WS performed the experiments. LC, YL and HQ analyzed the data. HQ and LC wrote the paper. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
No potential conflict of interest was reported by the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Identification of known miRNAs. Table S2 Identification of novel miRNAs.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Qu, H., Liu, Y., Jiang, H. et al. Identification and characterization of miRNAs associated with sterile flower buds in the tea plant based on small RNA sequencing. Hereditas 158, 26 (2021). https://doi.org/10.1186/s41065-021-00188-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41065-021-00188-8