
Abstract
Background
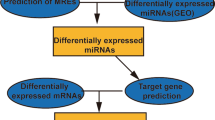
This study aimed to identify potential crucial genes and construction of microRNA-mRNA negative regulatory networks in osteosarcoma by comprehensive bioinformatics analysis.
Methods
Data of gene expression profiles (GSE28424) and miRNA expression profiles (GSE28423) were downloaded from GEO database. The differentially expressed genes (DEGs) and miRNAs (DEMIs) were obtained by R Bioconductor packages. Functional and enrichment analyses of selected genes were performed using DAVID database. Protein–protein interaction (PPI) network was constructed by STRING and visualized in Cytoscape. The relationships among the DEGs and module in PPI network were analyzed by plug-in NetworkAnalyzer and MCODE seperately. Through the TargetScan and comparing target genes with DEGs, the miRNA-mRNA regulation network was established.
Results
Totally 346 DEGs and 90 DEMIs were found to be differentially expressed. These DEGs were enriched in biological processes and KEGG pathway of inflammatory immune response. 25 genes in the PPI network were selected as hub genes. Top 10 hub genes were TYROBP, HLA-DRA, VWF, PPBP, SERPING1, HLA-DPA1, SERPINA1, KIF20A, FERMT3, HLA-E. PPI network of DEGs followed a pattern of power law network and met the characteristics of small-world network. MCODE analysis identified 4 clusters and the most significant cluster consisted of 11 nodes and 55 edges. SEPP1, CKS2, TCAP, BPI were identified as the seed genes in their own clusters, respectively. The miRNA-mRNA regulation network which was composed of 89 pairs was established. MiR-210 had the highest connectivity with 12 target genes. Among the predicted target of MiR-96, HLA-DPA1 and TYROBP were the hub genes.
Conclusion
Our study indicated possible differentially expressed genes and miRNA, and microRNA-mRNA negative regulatory networks in osteosarcoma by bioinformatics analysis, which may provide novel insights for unraveling pathogenesis of osteosarcoma.
Similar content being viewed by others


Background
Osteosarcoma is the most common primary malignant bone cancer in children and adolescents, which originates from mesenchymal stem cells and exhibits osteoblastic differentiation [1]. The incidence rate of osteosarcoma is approximately one to three cases per million each year worldwide [2]. With the development of surgery and chemotherapy, the survival rate in osteosarcoma patients without distal metastasis has been largely increased [3]. However, despite improvements in osteosarcoma therapy over the last three decades, the overall survival of patients has reached a plateau and about 30–40% of the patients experience progressive metastasis within 5 years after diagnosis and die [4]. Therefore, it is extremely necessary to explore novel biomarkers and therapeutic targets for osteosarcoma.
In recent years, the developments in molecular biology have provided some new insights into potential diagnostic and therapeutic biomarkers for osteosarcoma. For instance, it was found that SPARCL1 was downregulated in osteosarcoma by epigenetic methylation of promoter DNA and activating the expression of SPARCL1 could inhibit the osteosarcoma metastasis in vitro and in vivo [5]. NRP2 was reported to be up-regulated in osteosarcoma cell lines and tissues, and associated with poor survival of osteosarcoma patients [6]. On the other hand, numerous studies shows that microRNAs (miRNAs) may play essential roles in osteosarcoma tumorigenesis by negatively regulating expression level of target gene. MiR-497, for instance, can activate P21 expression by inhibiting the expression of MAPK/Erk signaling pathway, and promote the apoptosis of osteosarcoma cells [7]. However, the miRNA-mRNA negative regulation network in osteosarcoma had been not fully delineated by now.
In this study we used bioinformatics methods to integrate miRNA and mRNA expression data, which are available in the GEO database, to identify differentially expressed genes (DEGs) and miRNAs (DEMIs) between osteosarcoma and normal cell, and establish the miRNA-mRNA negative regulation network, aiming to provide valuable information for use in defining the mechanism of pathogenesis in osteosarcoma.
Methods
Identification of differentially expressed genes and miRNAs from public microarray data
To explore the DEGs and DEMIs in osteosarcoma compared to normal bone, the public gene expression (GSE28424) and miRNA expression (GSE28423) profiles were downloaded from the Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo). These profiles were deposited by Namløs HM et al. in 2011 and composed with the same cell lines: 19 OS cell lines (case group) and 4 normal bone samples (control group) [8]. Then, the dataset was analyzed by R Bioconductor packages and raw datasets were normalized based on the preprocess Core package and the DEGs and DEMIs were screened out via the limma package through the cut-off criteria of adjusted P-value< 0.01 and |Log2(FC)| > 2.
Functional and pathway enrichment analysis
The Database for Annotation, Visualization and Integrated Discovery (DAVID, https://david.ncifcrf.gov/) was used to perform functional and pathway enrichment analysis. DAVID is a systematic and integrative functional annotation tools which allows investigators to unravel biological meaning behind large list of genes [9]. Gene ontology (GO) analysis including the cellular component (CC), molecular function (MF), and biological process (BP) [10] and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis [11] were carried out for the DEGs. P < 0.05 was regarded as statistical significance.
Protein–protein interaction (PPI) network construction and module analysis
In order to interpret the molecular mechanisms of key cellular activities in osteosarcoma, The online tool, Search Tool for the Retrieval of Interacting Genes database (STRING), was used to construct PPI network of the DEGs [12]. The interaction score of not < 0.7 (high confidence score) was considered significant and the PPI was visualized.
The relationships among the DEGs were analyzed by plug-in NetworkAnalyzer of Cytoscape software for the characteristics of small-world network through calculating the network properties such as distribution of network node degree, distribution of the shortest path, average aggregation coefficient and proximity to the center [13]. Subsequently, the hub genes were selected according to connection degree. Moreover, Molecular Complex Detection (MCODE) was applied to find clusters of genes in PPI network. “Degree cutoff = 2”, “node score cutoff = 0.2”, “k-core = 2” and “max. depth = 100” were set as the cut-off criterion.
Prediction of miRNA targets
The target genes of DEMIs were predicted through the Targetscan (http://www.targetscan.org/), an online program that predicts targets of miRNAs by seeking the specific sequence complementary to the seed region of each miRNA. According to the predicted efficacy of targeting as calculated using cumulative weighted context++ scores of the sites, predicted targets are ranked. In this study, the genes with the cumulative weighted context++ scores ≤ − 0.4 were selected as target genes of each miRNA. Furthermore, we only selected the reverse pairs that contained the DEMIS and overlapping genes of DEGs. Finally, miRNA–mRNA negative regulatory network depicting interactions between miRNAs and their potential targets in osteosarcoma was visualized using Cytoscape.
Results
Identification of DEGs and DEMIs
Compared with normal bone samples, a total of 346 DEGs were identified in the osteosarcoma cells, which contained 43 up-regulated and 303 down-regulated genes. The top ten up-regulated and down-regulated genes are listed in Table 1. In total, 90 DEMIs were found to be differentially expressed. 58 DEMIs were downregulated and 32 DEMIs were upregulated. The top ten significantly differentially expressed miRNAs are showed in Table 2.
GO functional annotation and pathway enrichment
The top 10 significant terms of GO functional annotation and pathway enrichment analysis in DAVID were illustrated as Fig. 1.

Gene ontology and pathway enrichment analysis of the differentially expressed genes in osteosarcoma
In the CC ontology, we found that the majority of enriched categories were relevant to extracellular components, such as extracellular exosome(120 genes), extracellular space (77 genes), extracellular region (72 genes), and extracellular matrix (20 genes). The second majority of enriched categories were associated with cytosol (95 genes). In addition, the other enriched CC GO terms contained cell surface (25 genes), MHC class II protein complex (8 genes) and proteinaceous extracellular complex (18 genes). In the BP ontology, the regulation of inflammatory immune response items constitute the majority of enriched GO categories, including immune response (35 genes), innate immune response (26 genes), inflammatory response (26 genes), defense response to Gram-positive bacterium (12 genes), defense response(11 genes) and antigen processing and presentation (9 genes) and antigen processing and presentation of peptide or polysaccharide antigen via MHC class II (7 genes). The other enriched BP GO terms contained platelet degranulation (12 genes), muscle filament sliding (9 genes)and oxygen transport (6 genes). In the MF ontology, the binding-related items constitute the majority of enriched GO categories, including actin binding (17 genes), heparin binding (13 genes), heme binding (11 genes), protease binding (10 genes), collagen binding (9 genes), peptide antigen binding (7 genes) and oxygen binding (7 genes). Besides, the other enriched categories comprised items related to structural constituent of muscle (10 genes), oxygen transporter activity (6 genes) and MHC class II receptor activity (6 genes).
Furthermore, the KEGG pathways of DEGs mainly involved in inflammatory immune response, which included Phagosome (19 genes), Staphylococcus aureus infection (15 genes), Cell adhesion molecules (CAMs) (15 genes), Systemic lupus erythematosus (14 genes), Asthma (11 genes) and et al.
PPI network construction, module analysis and hub gene selection
PPI networks were constructed on the basis of STRING database and displayed in Fig. 2. We also analyzed the network properties, as shown in Fig. 3a–d. We could see the distribution of network node degrees followed a pattern of power law network in Fig. 3a, proximity to center in Fig. 3b, the average clustering coefficient in Fig. 3c and fromthe shortest path in Fig. 3d and we could also see that they meet the characteristics of small world network. When “Degree≥10” was set as the cut-off criterion, 25 genes in the PPI network were selected as hub genes in osteosarcoma. Top 10 hub genes were TYROBP, HLA-DRA, VWF, PPBP, SERPING1, HLA-DPA1, SERPINA1, KIF20A, FERMT3, HLA-E, and showed in Fig. 2. These hub genes might play crucial roles in osteosarcoma.

The protein-protein interaction network for the the differentially expressed genes and miRNA-target gene regulatory network in osteosarcoma. Circular nodes in light blue represent the differentially expressed genes; diamond nodes in yellow represent the hub genes; Circular nodes in red represent the screened out differentially expressed miRNAs and arrows in nodes represent the change trend of miRNAs

PPI network analysis of DEGs for characteristics of small-world network. (a) Distribution of degrees (b) the proximity to the centre (c) average aggregation coefficient (d) distribution of the shortest path
Subsequently, when “score ≥ 5” was defined as the cut-off criterion in MCODE, 4 clusters were identified from PPI network, and the most significant cluster consisted of 11 nodes and 55 edges. Furthermore, MCODE analysis showed that each cluster contained one seed gene. SEPP1 (one of the hub genes), CKS2 (the predicted target of MiR-513c), TCAP (one of the predicted target of MiR-18a), BPI (one of the predicted target of MiR-93) were identified as the seed genes in their own clusters, respectively [Fig. 4].

Four significant modules selected from protein-protein interaction network
miRNA-mRNA negative regulation network
Through the TargetScan, the target genes of 90 DEMIs were predicted. By comparing target genes with DEGs, we screened out 35 DEMIs (25 upregulated and 10 downregulated miRNA) and 78 DEGs (67downregulated and 11 upregulated mRNA), presented an expression trend opposite to each other in osteosarcoma. And then, the miRNA-mRNA regulation network which was composed of 89 pairs was established and shown in Fig. 2. In our study, miR-9, the most significantly upregulated among these screened out DEMIS, was predicted to 2 downregulated target, including CEACAM6 and PDK4. MiR-210 was with the highest connectivity with target genes, targeting 12 differentially expressed genes (ANGPTL4, AQP1, ARHGAP25, BTG2, CD247, DNASE1L3, LYL1, P2RY8, SH2D3C, SRL, STAT5A, TNFRSF1B), that might indicate its important role in osteosarcoma. Among the predicted target of MiR-96, HLA-DPA1 and TYROBP were the hub genes. Moreover, several genes were predicted to the common targets of different miRNA. For instance, CSF1R, one of the hub genes, was predicted as the common target of miR-149 and miR-22.
Discussion
Unraveling the mechanisms of the initiation and development of osteosarcoma would benefit the diagnosis, treatment and prognosis evaluation. In present study, two types of microarray datasets: gene expression profile and miRNA expression profile were downloaded from the GEO and analyzed for their relationship in osteosarcoma. Compared with normal bone samples, a total of 346 DEGs were identified in the osteosarcoma cells, containing 43 up-regulated and 303 down-regulated genes. CBS (cystathionine-β-synthase cancer), the most regulated gene in this study, was found to be upregulated in many types of tumors, such as multiple myeloma [14], colorectal cancers [15], bladder cancer [16] and et al., and downregulation of CBS help inhibit carcinogenesis. However, to our knowledge, there was no report about CBS in osteosarcoma, so it might be a novel target for the diagnosis or treatment of osteosarcoma.
As was suggested by DAVID analysis, the DEGs in osteosarcoma were enriched in biological processes and KEGG pathway of inflammatory immune response. Molecular function of GO analysis showed enrichment in binding-related items. It is reasonable because immune system has significant roles in tumorigenesis. At the cancer initiation stage, the immune system can recognize tumors expressing foreign antigens and eradicate tumors via innate and adaptive immune activation [17]. If tumor cells are not eliminated via recognition by the immune system, tumor cells can enter an equilibrium phase and evolve a number of phenotypic changes and dampen immunogenicity to escape the immune surveillance [18, 19]. In other words the immune microenvironment helps cancer cells to select the dominant cells so that the tumor can progressively grow unhindered. Therefore, osteosarcoma tumors are infiltrated with immune cells that may have the ability to fight tumor cells [20] but are tolerized due to immune escape of the tumor cells [21, 22]. Cellular component of GO analysis showed the majority of enriched categories were relevant to extracellular components, such as extracellular exosome, extracellular space, extracellular region, and extracellular matrix. Tumor microenvironment has complementary effects on the development and metastasis of osteosarcoma through extracellular secretion, alteration of phenotype type of tumor cells, immune escape and providing proper acid-base environment for tumor cells [23].
PPI network of DEGs illustrated the overview of their functional connections, which followed a pattern of power law network from the shortest path, average aggregation coefficient, node degree and proximity to the center and met the characteristics of small-world network, and of which 25 hub genes were selected. Most of them were enriched in inflammatory immune response and platelet degranulation. As we known, the activated degranulated platelet accumulated on the surface of tumor cell to protect them from the immune system recognization and eradication. After module analysis of the PPI network, 4 seed genes were selected, such as SEPP1, CKS2, TCAP and BPI genes.
SEPP1, a major selenium transport protein, has endogenous antioxidant function through catalyzing the oxidation of glutathione by a hydrogen peroxide or phosphatidylcholine hydroperoxide [24]. It was found that SEPP1 levels and activity are significantly decreased in colon tumors, human prostate tumors, C3(1)/Tag transgenic ouse tumors, and prostate cancer cell lines [25, 26]. Furthermore, several single nucleotide polymorphisms (SNPs) have been identified in SEPP1 that may contribute to decreased expression in colorectal adenomas and have been associated with cancer risk [27, 28]. CKS2 (Cyclin-dependent kinases regulatory subunit 2), a cyclin-dependent kinase interacting protein, is essential for cell cycle regulation. Elevated expression of CKS2 has been demonstrated in multiple types of human malignancies. In prostate cancer, aberrant expression of CKS2 contributes to tumorigenesis by enhancing cell proliferation and inhibiting programmed cell death [29]. In a papillary thyroid carcinoma, miR-26a [30] and miR-7 [31] modulates tumor growth and tumorigenesis by targeting CKS2. In colorectal cancer cells, attenuation of CKS2 results in decreased cell viability, increased cell apoptosis and cell cycle arrest [32]. In almost the same way, aberrant expression of TCAP and BPI was in relation with multiple types of tumor. However, no study has associated these seed genes with osteosarcoma, which might indicate them as the new targets.
It has been shown that miRNAs induce RNA silencing by targeting 3’-UTR of mRNAs, and that miRNA functions as oncogenes or oncosuppressor depending on the function of suppressed targets. In present study, the miRNA-mRNA regulation network which was composed of 89 pairs was established through the TargetScan and comparing target genes with DEGs, in which MiR-210 directly regulated 12 differentially expressed genes and was significantly upregulated in osteosarcoma. Mounting evidence identified MiR-210 as an oncogenic role in generating osteosarcoma; increased expression of MiR-210 is associated with decreased overall survival and progression-free survival, and more frequently occurred in osteosarcoma tissues with large tumor size, poor response to preoperative chemotherapy, and positive metastasis [33, 34]. Nonetheless, the defined roles of miR-210 in osteosarcoma malignant progression, especially the target gene of miR-210 in osteosarcoma dedifferentiation, was insufficiently researched. Only NFIC, a validated target of miR-210 from miRBase and a DEG from a gene expression profile GSE38135, had been shown to play important role in TGF-b1-induced osteosarcoma dedifferentiation and can be significantly reduced by miR-210 treatment in human osteosarcoma cell line MNNG/HOS [35]. So far, the role of other miR-210-gene pairs in osteosarcoma was not reported. HLA-DPA1 and TYROBP, the hub genes in PPI network were regulated by MiR-96, but their function were not researched in osteosarcoma too. These might imply that miRNA and their target genes may represent potential novel therapeutic targets or biomarkers for osteosarcoma.
The limitation in our work is that the pathogenesis of key miRNA and gene in osteosarcoma need to be elucidated through experiments in vivo and in vitro.
Conclusion
In summary, our study was intended to identify key genes in osteosarcoma and construct regulatory networks between miRNA and mRNA through bioinformatics analysis. 25 hub genes and 4 seed genes were identified according to PPI network.
Functional and pathway enrichment analysis indicated immune system played significant roles in osteosarcoma tumorigenesis. Additionally, deregulated miRNAs such as MiR-210 and MiR-96, might exert their biological functions through their targeting mRNAs. Moreover, our results could provide novel sights in the mechanisms of the initiation and development of osteosarcoma, that would become the new diagnostic biomarkers and treatment targets for osteosarcoma.
Abbreviations
- BP:
-
Biological processes
- CC:
-
Cell component
- DEG:
-
Differentially expressed gene
- DEMI:
-
Differentially expressed miRNAs
- GO:
-
Gene ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- MF:
-
Molecular function
- PPI:
-
Protein–protein interaction
- STRING:
-
Search Tool for the Retrieval of Interacting Genes
- TCGA:
-
The Cancer Genome Atlas
References
Durfee RA, Mohammed M, Luu HH. Review of osteosarcoma and current management. Rheumatol Ther. 2016;3:221–43.
Jo VY, Fletcher CD. WHO classification of soft tissue tumours: an update based on the 2013 (4th) edition. Pathology. 2014;46:95–104.
Anderson ME. Update on survival in osteosarcoma. Orthop Clin North Am. 2016;47:283–92.
Lewis IJ, Nooij MA, Whelan J, Sydes MR, Grimer R, Hogendoorn PC, Memon MA, Weeden S, Uscinska BM, van Glabbeke M, et al. Improvement in histologic response but not survival in osteosarcoma patients treated with intensified chemotherapy: a randomized phase III trial of the European osteosarcoma intergroup. J Natl Cancer Inst. 2007;99:112–28.
Zhao SJ, Jiang YQ, Xu NW, Li Q, Zhang Q, Wang SY, Li J, Wang YH, Zhang YL, Jiang SH, et al. SPARCL1 suppresses osteosarcoma metastasis and recruits macrophages by activation of canonical WNT/β-catenin signaling through stabilization of the WNT-receptor complex. Oncogene. 2018;37:1049–61.
Ji T, Guo Y, Kim K, McQueen P, Ghaffar S, Christ A, Lin C, Eskander R, Zi X, Hoang BH. Neuropilin-2 expression is inhibited by secreted Wnt antagonists and its down-regulation is associated with reduced tumor growth and metastasis in osteosarcoma. Mol Cancer. 2015;14:86.
Gui ZL, Wu TL, Zhao GC, Lin ZX, Xu HG. MicroRNA-497 suppress osteosarcoma by targeting MAPK/Erk pathway. Bratisl Lek Listy. 2017;118:449–52.
Namløs HM, Meza-Zepeda LA, Barøy T, Østensen IH, Kresse SH, Kuijjer ML, Serra M, Bürger H, Cleton-Jansen AM, Myklebost O. Modulation of the osteosarcoma expression phenotype by microRNAs. PLoS One. 2012;7(10):e48086.
Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:P3.
Gene Ontology Consortium. The gene ontology (GO) project in 2006. Nucleic Acids Res. 2006;34(Database issue):D322–6.
Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30.
Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, von Mering C. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(Database issue):D447–52.
Assenov Y, Ramírez F, Schelhorn SE, Lengauer T, Albrecht M. Computing topological parameters of biological networks. Bioinformatics. 2008;24(2):282–4.
De Vos J, Thykjaer T, Tarte K, Ensslen M, Raynaud P, Requirand G, Pellet F, Pantesco V, Rème T, Jourdan M, et al. Comparison of gene expression profiling between malignant and normal plasma cells with oligonucleotide arrays. Oncogene. 2002;21:6848–57.
Phillips CM, Zatarain JR, Nicholls ME, Porter C, Widen SG, Thanki K, Johnson P, Jawad MU, Moyer MP, Randall JW, et al. Upregulation of cystathionine-β-synthase in colonic epithelia reprograms metabolism and promotes carcinogenesis. Cancer Res. 2017;77:5741–54.
Gai JW, Qin W, Liu M, Wang HF, Zhang M, Li M, Zhou WH, Ma QT, Liu GM, Song WH, et al. Expression profile of hydrogen sulfide and its synthases correlates with tumor stage and grade in urothelial cell carcinoma of bladder. Urol Oncol. 2016;34:166.e15–20.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–60.
Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, Smyth MJ, Schreiber RD. Adaptive immunity maintains occult cancer in an equilibrium state. Nature. 2007;450:903–7.
Théoleyre S, Mori K, Cherrier B, Passuti N, Gouin F, Rédini F. Phenotypic and functional analysis of lymphocytes infiltrating osteolytic tumors: use as a possible therapeutic approach of osteosarcoma. BMC Cancer. 2005;5:123.
Laborde RR, Lin Y, Gustafson MP, Bulur PA, Dietz AB. Cancer vaccines in the world of immune suppressive monocytes (CD14(+)HLA-DR(lo/neg) cells): the gateway to improved responses. Front Immunol. 2014;5:147.
Kawano M, Itonaga I, Iwasaki T, Tsumura H. Enhancement of antitumor immunity by combining anti-cytotoxic T lymphocyte antigen-4 antibodies and cryotreated tumor lysate-pulsed dendritic cells in murine osteosarcoma. Oncol Rep. 2013;29:1001–6.
Wang M, Zhao J, Zhang L, Wei F, Lian Y, Wu Y, Gong Z, Zhang S, Zhou J, Cao K, et al. Role of tumor microenvironment in tumorigenesis.J. Cancer. 2017;8:761–73.
Barrett CW, Short SP, Williams CS. Selenoproteins and oxidative stress-induced inflammatory tumorigenesis in the gut. Cell Mol Life Sci. 2017;74:607–16.
Al-Taie OH, Uceyler N, Eubner U, Jakob F, Mörk H, Scheurlen M, Brigelius-Flohe R, Schöttker K, Abel J, Thalheimer A, et al. Expression profiling and genetic alterations of the selenoproteins GI-GPx and SePP in colorectal carcinogenesis. Nutr Cancer. 2004;48:6–14.
Calvo A, Xiao N, Kang J, Best CJ, Leiva I, Emmert-Buck MR, Jorcyk C, Green JE. Alterations in gene expression profiles during prostate cancer progression: functional correlations to tumorigenicity and down-regulation of selenoprotein-P in mouse and human tumors. Cancer Res. 2002;62:5325–35.
Steinbrecher A, Méplan C, Hesketh J, Schomburg L, Endermann T, Jansen E, Akesson B, Rohrmann S, Linseisen J. Effects of selenium status and polymorphisms in selenoprotein genes on prostate cancer risk in a prospective study of European men. Cancer Epidemiol Biomark Prev. 2010;19:2958–68.
Peters U, Chatterjee N, Hayes RB, Schoen RE, Wang Y, Chanock SJ, Foster CB. Variation in the selenoenzyme genes and risk of advanced distal colorectal adenoma. Cancer Epidemiol Biomark Prev. 2008;17:1144–54.
Lan Y, Zhang Y, Wang J, Lin C, Ittmann MM, Wang F. Aberrant expression of Cks1 and Cks2 contributes to prostate tumorigenesis by promoting proliferation and inhibiting programmed cell death. Int J Cancer. 2008;123:543–51.
Lv M, Zhang X, Li M, Chen Q, Ye M, Liang W, Ding L, Cai H, Fu D, Lv Z. miR-26a and its target CKS2 modulate cell growth and tumorigenesis of papillary thyroid carcinoma. PLoS One. 2013;8:e67591.
Hua K, Jin J, Zhang H, Zhao B, Wu C, Xu H, Fang L. MicroRNA-7 inhibits proliferation, migration and invasion of thyroid papillary cancer cells via targeting CKS2. Int J Oncol. 2016;49:1531–40.
Yu MH, Luo Y, Qin SL, Wang ZS, Mu YF, Zhong M. Up-regulated CKS2 promotes tumor progression and predicts a poor prognosis in human colorectal cancer. Am J Cancer Res. 2015;5:2708–18.
Cai H, Lin L, Cai H, Tang M, Wang Z. Prognostic evaluation of microRNA-210 expression in pediatric osteosarcoma. Med Oncol. 2013;30:499.
Wang J, Zhao J, Shi M, Ding Y, Sun H, Yuan F, Zou Z. Elevated expression of miR-210 predicts poor survival of cancer patients: a systematic review and meta-analysis. PLoS One. 2014;9:e89223.
Zhang H, Mai Q, Chen J. MicroRNA-210 is increased and it is required for dedifferentiation of osteosarcoma cell line. Cell Biol Int. 2017;41:267–75.
Funding
This research was supported by Science and Technology Plan of Xiamen (3502Z20154067).
Availability of data and materials
The authors declare that the data supporting the findings of this study are available within the article.
Author information
Authors and Affiliations
Contributions
YP collected the data; LL and ZY performed the statistical analysis; JC wrote the paper; ZD conceived the study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Pan, Y., Lu, L., Chen, J. et al. Identification of potential crucial genes and construction of microRNA-mRNA negative regulatory networks in osteosarcoma. Hereditas 155, 21 (2018). https://doi.org/10.1186/s41065-018-0061-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41065-018-0061-9