
Abstract
The discovery that several demyelinating disorders in children may be associated with autoantibodies to astrocytes, myelin, and/or synaptic proteins has opened the possibility of their use as diagnostic and prognostic biomarkers. The identification of aquaporin 4 (AQP4) antibodies as specific diagnostic markers of neuromyelitis optica (NMO) led to an expansion of the clinical spectrum of this disorder to include patients with incomplete forms or with manifestations outside of the optic nerves and spinal cord. Recently, an international panel of experts proposed the use of the term NMO spectrum disorders (NMOSD) to encompass all of these phenotypes. Although rare in children, the early identification of AQP4 antibodies has important prognostic and therapeutic implications as their presence is highly predictive of relapses and accumulation of disability. More recently, antibodies to myelin oligodendrocyte glycoprotein (MOG) have been described in children and adults diagnosed with NMOSD and seronegative for AQP4 antibodies, as well as in children with other acquired demyelinating disorders. For example 60 % of children with acute demyelinating encephalomyelitis (ADEM) have MOG antibodies. Whether MOG antibodies are pathogenic or surrogate biomarkers of the disease is unclear but their identification is important for two reasons. First, patients with a first demyelinating event who are MOG antibody positive are unlikely to develop multiple sclerosis (MS); and second, the long-term persistence of MOG antibodies associates with recurrent non-MS demyelinating disorders such as NMOSD, recurrent optic neuritis or transverse myelitis, or multiphasic ADEM. Other antibodies, such as those targeting Kir 4.1 or the glycine receptor have also been described in association with pediatric MS and other demyelinating disorders but their clinical relevance, and existence, in the case of Kir 4.1, is still under investigation. The focus of this review will be the current data on the antibodies mentioned above, and the recent discovery that some children have both a demyelinating disorder and autoimmune encephalitis. The presence of antibodies against targets related to demyelinating disorders (AQP4 or MOG) and those related to autoimmune encephalitis (e.g., N-methyl-D-aspartate receptor), supports the concurrence of two autoimmune disorders in these patients.
Similar content being viewed by others
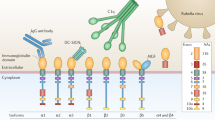

Background
To establish the correct diagnosis and prognosis after a first demyelinating episode in a child is challenging as, at this age, different acquired demyelinating disorders (ADS) can present with similar clinical features. It can be difficult to distinguish between a monophasic illness such as acute demyelinating encephalomyelitis (ADEM), which will not require long-term treatment, and the first episode of a chronic or relapsing disorder such as multiple sclerosis (MS), neuromyelitis optica spectrum disorders (NMOSD), or multiphasic ADEM, in which early initiation of treatment can be crucial for preventing long-term disability [1]. Brain and spinal cord MRI as well as the evaluation for the presence of oligoclonal bands in the cerebrospinal fluid (CSF), are well-characterized tools that can assist in formulating the diagnosis of ADS [2, 3]. More recently, the discovery that several ADS may be associated with autoantibodies to astrocytes, myelin, and/or synaptic proteins opens the possibility of utilizing these as biomarkers for diagnosis, prognosis and choice of appropriate therapies [4–6]. Here we review the current knowledge about the antibodies associated with ADS in children. The demographic, clinical and laboratory information according to the antibody is summarized in Table 1.

MRI in children with demyelinating syndromes associated with MOG or AQP4 antibodies. (a, d): Brain and spinal cord MRI of a 13-year old girl with NMOSD and AQP4 antibodies. The sagittal MRI of the spinal cord shows hyperintensity involving more than 3 segments (a) whereas the brain axial T2-sequence shown in (d) is normal. (b, c, e, f): Coronal FLAIR-sequences of an 8-year old girl with an initial episode of ADEM and MOG antibodies showing bilateral widespread subcortical lesions (b, c). Seven months after the attack there has been complete resolution of the lesions (e, f)
Antibodies to astrocytes or myelin proteins
Anti-Aquaporin 4 (AQP4) antibodies and NMOSD
Neuromyelitis optica (NMO) is a rare disease. Population-based studies estimate a prevalence of 0.52–4.4 cases/100,000 inhabitants, with differences across studies according to geographic and ethnic cohorts [7]. NMO is characterized by recurrent inflammatory episodes that preferentially affect the optic nerves and spinal cord [8]. The disorder predominantly affects women (90 %), and although the median age at onset is 39 years, patients of all ages can be affected including children and the elderly [9]. Overall, up to 90 % of patients have a recurrent form of the disease, frequently with incomplete recovery between relapses with incremental disability [10, 11]. Although NMO was initially considered to be an optical-spinal form of MS, today it is accepted as a distinct clinical-pathological entity. The underlying pathophysiology was suspected to be B-cell mediated due to distinctive pathologic findings showing vasculocentric deposition of immunoglobulins (Ig), complement components within lesions, and predominance of neutrophils and eosinophils in the inflammatory infiltrates that are typical features of a type-2 T-helper cell immune response [12]. The involvement of the humoral immune system in NMO was confirmed by Lennon and colleagues with the identification of antibodies against AQP4, a water channel expressed predominantly in astrocyte end-feet [4, 13]. Subsequent studies demonstrated that AQP4 antibodies, which belong to the IgG1 subclass, activate complement and disrupt the AQP4 water channel, disturbing water homeostasis, promoting edema and perivascular inflammation [14, 15]. The pathogenic role of AQP4 antibodies in NMO has been demonstrated in murine models showing NMO-like lesions after passive transfer of AQP4 antibodies from human patients [15, 16].
The identification of AQP4 antibodies led in 2006 to revised NMO criteria [8]. In 2007 the term NMOSD was introduced to include AQP4 seropositive patients with limited or inaugural forms (e.g., recurrent optic neuritis or longitudinally extensive transverse myelitis [LETM]), and those with manifestations outside of the optic nerves and spinal cord (e.g., intractable hiccups or vomiting), secondary to involvement of areas also known to be rich in AQP4 such as periventricular areas of the third and fourth ventricles, and periaqueductal and parahippocampus grey matter [17–19]. Most recently, an International Panel proposed new diagnostic criteria that unified under the term NMOSD traditional NMO and modern NMOSD definitions [20]. The main reasons for the unifying term were based on the assumption that there are no biological differences between patients diagnosed with NMO and those with NMOSD in AQP4 seropositive patients, and that patients with limited forms frequently develop subsequent clinical attacks consistent with conventional NMO in AQP4 seropositive patients [20]. For example, in a comparative study of patients with AQP4 antibodies and NMO and patients with limited forms, we found no differences in motor or visual disability outcome, supporting the use of the term NMOSD for patients with AQP4 antibodies [21]. However, the 2015 criteria allow the diagnosis of NMOSD in patients without AQP4, incorporating these patients into the same group as those with AQP4, and this is controversial. For example, there are studies that have shown some clinical differences between AQP4 positive and negative patients including a lower female predominance in those without AQP4-IgG [22, 23]. The phenotypic variability between AQP4 positive and negative patients may be partially explained by the presence of antibodies to myelin oligodendrocyte glycoprotein (MOG) in a subgroup of AQP4 seronegative patients, as patients with MOG antibodies may develop a clinical picture indistinguishable from NMOSD but with better outcome [22, 24]. This likely represents a different immunopathogenic mechanism that would not be clear using descriptive clinical terms, such as NMO or NMOSD [24]. Therefore, some authors suggest replacing these terms by a pathogenically descriptive name such as “autoimmune channelopathy due to AQP4 antibodies” to exclude patients without AQP4 antibodies such as patients with MOG antibodies, which probably underly a different biologic disease. Autoimmune channelopathy due to AQP4 antibodies would also encompass other AQP4 related non-CNS disorders such as the recently described disorders with hyperCKemia and/or skeletal muscle involvement [25, 26]. This systemic involvement is not surprising as AQP4 channels are also expressed in normal muscles [27].
Characteristics of NMOSD with AQP4 antibodies in children
It is estimated that almost 70 % of adults with NMOSD have AQP4 antibodies [28]. In contrast these antibodies are rare in the pediatric population [29]. Among different prospective case series, up to 5 % of children with demyelinating diseases harbor these antibodies [30, 31]. However, a study evaluating specific clinical phenotypes found that almost half of children with classic NMO features harbored AQP4 antibodies [30].
The presence of these antibodies has important clinical and prognostic implications. For example, seropositivity for AQP4 antibodies in a first inflammatory episode is highly predictive for relapses, and relapses are associated with long-term disability in both children and adults [9, 29, 30]. In a series of 48 children with NMOSD and AQP4 antibodies, 54 % had visual impairment (27 % bilateral visual acuity ≤ 20/200), 54 % had residual weakness (29 % with paraparesis), and 12 % had sphincter involvement at last follow-up (median 12 months) [29].
Another example of the importance of identifying these antibodies is that in children AQP4 antibody associated disorders can present with encephalopathy mimicking ADEM or with lesions on brain MRI that simulate MS [1, 29, 32, 33]. Moreover, CSF abnormalities in pediatric MS may mimic those ordinarily considered suggestive of NMOSD [29, 30, 33]. Additionally, while an adult with LETM is commonly suspected to have AQP4-NMOSD, in children, this diagnosis is often not suspected as LETM also occurs with ADEM and MS [1]. Other differences between children and adults with NMOSD appears to be a lower female preponderance in children (3:1 female:male ratio compared with up to 9:1 for adults) [33]; and that a greater proportion of children may have monophasic disease [30, 33]. Aside from these caveats, the currently proposed diagnostic criteria for NMOSD are considered appropriate for pediatric patients [20].
Misdiagnosing NMOSD for MS in children can be deleterious as standard treatments for MS such as beta interferon, fingolimod or natalizumab can result in worsening of NMOSD [34, 35]. In contrast, treatment strategies that selectively target the cellular and soluble components of the humoral immune response in NMOSD, such as plasma exchange and B-cell depletion (e.g., rituximab), have been shown to be effective in the prevention of NMOSD relapses [36].
Based in the growing knowledge about the pathophysiology of NMOSD with AQP4 antibodies there are many studies examining new targeted therapies that will hopefully result in better outcomes for these patients. Examples include eculizumab, which acts to inhibit complement activation; tocilizumab, which acts by blocking the cytokine IL6 receptor; antibodies directed against the CD19 molecule expressed by B cells; and the monoclonal antibody aquaporimab, which is designed to block the interaction between human IgG and AQP4 [37–40].
In summary, for the reasons noted above although the prevalence of AQP4 antibodies is low in the pediatric population, determination of these antibodies should be considered in any child with a first demyelinating episode involving one or more areas rich in AQP4.
MOG antibody-associated disorders in children
Antibodies against MOG have been associated with several types of acquired demyelinating disorders in children [5]. The specificity of MOG antibodies for demyelinating disorders was questioned in the past due to conflicting results from studies using ELISA or western blot, techniques that measure antibody binding to linear epitopes. More recently, the use of CBA (cell-based assays), in which antibody binding to conformational epitopes is assessed, has shown that MOG antibodies are able to identify patients with acquired demyelinating disorders [5]. These findings demonstrate the value of a multi-technique laboratory approach in autoantibody research and validation. Although there is some in vitro and in vivo data suggesting a pathogenic role of MOG antibodies, [41] there are no studies demonstrating that passive transfer of these antibodies leads to clinical disease, as has been demonstrated in animal models of AQP4 autoimmunity [16].
Whether MOG antibodies are pathogenic or represent surrogate biomarkers of the disease is currently unclear. However, preliminary data support that the determination of these antibodies is important [5]. In children, ADEM is the clinical syndrome that most frequently associates with MOG antibodies (present in up to 60 % of cases) [5]. A recent study compared 19 children with ADEM with MOG antibodies to 14 patients with ADEM without these antibodies [42]. There were no differences in in age at presentation, gender, symptoms, initial disease severity, or the presence of oligoclonal bands between children with or without MOG antibodies. However, children with ADEM and MOG antibodies more often had MRI findings characterized by large, hazy, bilateral lesions in the brain and an increased frequency of LETM. Children with MOG also had more favorable outcomes compared to those lacking these antibodies [42].
Several studies have shown that MOG antibody titers decline and become negative over time in patients with monophasic ADEM [43, 44]. A few initial reports postulated that the long-term persistence of MOG antibodies in a child could be predictive of the eventual development of pediatric MS [43]. Recent studies demonstrate that in fact, MOG antibodies are rarely found in pediatric MS and their presence supports a diagnosis other than MS [45]. For example, in a recent prospective series of 65 children with a first demyelinating attack, only 2/23 (9 %) with MOG antibodies compared with 16/42 (38 %) without these antibodies were diagnosed with MS after one year of follow-up, supporting the positive predictive value of MOG antibodies for a non-MS disease course [45]. However as these studies are based on short follow-up periods, longer time is needed to exclude eventual conversion to MS.
In contrast, there are several studies in children that support an association of the long-term persistence of MOG antibodies with recurrent non-MS demyelinating disorders such as NMOSD, recurrent optic neuritis, transverse myelitis or multi-phasic ADEM (Armangue personal observation) [41, 46–48]. Interestingly, these antibodies have also been described in a subgroup of ADS patients who initially presented with monophasic ADEM that fulfilled all clinical and neuroradiological criteria who subsequently develop one or more episodes of optic neuritis (ON) without further neuroradiological changes suggestive of demyelination. This clinical scenario named by some authors as ADEM-ON, [49] does not fall into any of the current diagnostic categories of ADS in children proposed by the International Pediatric MS Study Group, [1] but these patients who are at risk for subsequent clinical events could have NMOSD, chronic relapsing inflammatory optic neuropathy or MS, require close follow-up.
After ADEM, the most frequent syndrome associated with MOG antibodies in children is NMOSD (20 % of patients) [5, 50]. Compared to NMOSD with or without AQP4 antibodies, NMOSD with MOG antibodies has distinctive clinical and prognostic features [22, 51, 52]. Patients with NMOSD and MOG antibodies are slightly more likely to be male in contrast to the greater female predominance in cases associated with AQP4. Patients with NMOSD with MOG are more likely to have optic nerve than spinal cord involvement as well as more frequent involvement of the conus and the basal ganglia on MRI, in contrast to what is seen in NMOSD AQP4 patients [22, 51, 52].
In adults, the presence of MOG is predictive of better outcome in NMOSD compared to NMOSD with AQP4 antibodie [22, 51, 52]. In a study in which we evaluated 174 adults patients with NMOSD, ON or transverse myelitis, patients with MOG antibodies more frequently had a monophasic course (41 % vs 7 %) and better prognosis (median EDSS score 1.5 vs 4.0) compared to patients with AQP4 antibodies, despite having similar severity scores at disease onset [22]. Whether these results can be extrapolated to the pediatric population is unknown and requires further study.
Pathological studies are not available from patients with NMOSD and MOG antibodies. Studies of CSF from these patients show increased levels of myelin basic protein and undetectable levels of glial fibrillary acidic protein (GFAP). This is in contrast to the CSF findings in patients with NMOSD and AQP4 antibodies that show low levels of myelin basic protein and elevated GFAP [53]. This supports the hypothesis that MOG associated disease is due to demyelinating pathology rather than an astrocytopathy, and this could explain the clinical differences between NMOSD patients with AQP4 and those with MOG antibodies.
Kir 4.1 IgG antibodies
In 2012, the KIR4.1 (also known as KCNJ10), a glial inwardly rectifying potassium channel expressed on oligodendrocytes and astrocytes, was proposed as a putative MS autoantigen. Using ELISA, the investigators reported KIR4.1-channel-specific IgG in serum of 47 % (189/397) of adult MS patients and < 1 % of patients with other neurological disorders [54]. In a subsequent study these antibodies were also reported in the serum of 57 % (24/47) of children with acquired demyelinating syndromes [55]. However this high prevalence of anti-KIR4.1 antibodies in patients with MS has not been corroborated by other groups. Using similar methods, other studies in MS patients have shown much lower frequencies of these antibodies (from < 1 to 26 %), and the antibodies were also present to the same degree in normal subjects and other disease controls such as NMOSD patients [56–58]. Moreover, a recent study using a multi-technique approach did not detect KIR4.1-specific IgG in serum or CSF from patients with MS. No KIR4.1 loss from glia was seen in MS lesions and false positive results were demonstrated on ELISA [56]. The discrepancies between the results of these studies are difficult to explain, [59] but different analytical approaches might, in part, explain the contradictory findings. The potential relationship between KIR4.1 antibodies in MS deserves further investigation [59].
Antibodies to the Neuronal Cell Surface
Overlap between demyelinating syndromes and autoimmune encephalitis
The differential diagnosis of encephalopathy in children includes ADEM, NMOSD and autoimmune encephalitis associated with antibodies to neuronal cell-surface or synaptic proteins such as N-methyl-D-aspartate receptor (NMDAR) and others [60]. The clinical syndrome of anti-NMDAR encephalitis is highly predictable in adults and teenagers and is characterized by the onset of psychiatric and behavioral problems that progress to decreased level of consciousness, seizures, dyskinesias, choreathetoid movements or postures, and breathing or autonomic instability [61, 62]. In young children, the syndrome is similar, but children more frequently present with seizures or abnormal movements. Moreover, behavioral changes are often misdiagnosed as they may include new onset temper tantrums or agitation, which are frequent and non-specific at this age [62, 63]. In contrast to what is found in ADEM or in other ADS, in anti-NMDAR encephalitis brain MRI is typically normal or may show non-specific FLAIR/T2 changes, sometimes with transient cortical-meningeal enhancement [64]. A new challenge that has emerged is the finding that patients may have both demyelinating disease and autoimmune encephalitis [6]. The presence of two immune disorders in these patients is supported by the finding of antibodies against targets related to demyelinating disorders (AQP4 or MOG) and those related to autoimmune encephalitis (e.g., NMDAR) [6]. In these patients, the two disorders may occur sequentially as independent episodes separated by weeks to years or may occur simultaneously [6, 65]. The latter is the most diagnostically challenging. The presence of clinical or radiologic features characteristic of demyelination in a patient with autoimmune encephalitis and neuronal cell-surface antibodies should prompt testing for MOG and/or AQP4 antibodies. Similarly, patients with demyelinating disorders who develop unusual symptoms such as dyskinesias, refractory seizures, or prominent psychiatric symptoms should be tested for cell-surface antibodies (preferably in CSF, see Table 1) [6, 66]. While the identification of different antibodies can confirm the dual diagnoses, awareness that these disorders may overlap is the first step to establishing the correct diagnoses.
Glycine antibodies and demyelinating syndromes
Antibodies to glycine receptor (GlyR) were initially detected in serum and CSF of patients with progressive encephalomyelitis with rigidity and myoclonus (PERM), [67] but have since been reported in other subgroups of patients including patients with stiff-person syndrome, cerebellar ataxia and several types of demyelinating disorders in children and adults (e.g., optic neuritis and MS) [31, 68, 69].
This lack of syndrome specificity and the fact these antibodies are frequently found in patients with ADS concurrent with other more pathogenically relevant antibodies such as MOG and/or AQP4, [69] suggests that these antibodies may be non-specific markers of autoimmunity.
Conclusions
The differential diagnoses of a child presenting with a first episode of demyelination is wide and includes diseases with different pathogenic mechanisms. This can delay diagnosis and initiation of appropriate therapies, which may affect disease course and outcome.
The discovery of AQP4 antibodies allows for early identification of patients with similar pathophysiology and prognosis, thereby supporting the use of specifically directed therapeutic interventions. Antibodies to MOG have recently been postulated to be markers of non-MS demyelinating diseases in children and may identify patients with high risk of developing relapses. The identification of AQP4 or MOG antibodies concurrent with NMDAR antibodies in patients with autoimmune encephalitis has uncovered the fact that some patients can have both autoimmune encephalitis and a demyelinating disorder. Further investigation will clarify the role of the antibodies discussed above in these diseases and will likely lead to the discovery of additional antibody biomarkers that may facilitate diagnosis and serve as prognostic tools.
Abbreviations
- ADEM:
-
acute demyelinating encephalomyelitis
- AQP4:
-
aquaporin-4
- CSF:
-
cerebrospinal fluid
- GFAP:
-
glial fibrillary acidic protein
- GlyR:
-
glycine receptor
- Ig:
-
immunoglobulins
- LETM:
-
longitudinally extensive transverse myelitis
- MOG:
-
myelin oligodendrocyte glycoprotein
- MS:
-
multiple sclerosis
- NMDAR:
-
N-methyl-D-aspartate-receptor
- NMO:
-
neuromyelitis optica
- NMOSD:
-
neuromyelitis optica spectrum disorders
- ON:
-
optic neuritis
- PERM:
-
progressive encephalomyelitis with rigidity and myoclonus
References
Krupp LB, Tardieu M, Amato MP, et al. International Pediatric Multiple Sclerosis Study Group criteria for pediatric multiple sclerosis and immune-mediated central nervous system demyelinating disorders: revisions to the 2007 definitions. Mult Scler. 2013;19:1261–7.
Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69:292–302.
Heussinger N, Kontopantelis E, Gburek-Augustat J, et al. Oligoclonal bands predict multiple sclerosis in children with optic neuritis. Ann Neurol. 2015;77:1076–82.
Lennon PVA, Wingerchuk DM, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: Distinction from multiple sclerosis. Lancet. 2004;364:2106–12.
Reindl M, Di Pauli F, Rostásy K, Berger T. The spectrum of MOG autoantibody-associated demyelinating diseases. Nat Rev Neurol. 2013;9:455–61.
Titulaer MJ, Höftberger R, Iizuka T, et al. Overlapping demyelinating syndromes and anti-N-methyl-D-aspartate receptor encephalitis. AnnNeurol. 2014;75:411–28.
Pandit L, Asgari N, Apiwattanakul M, et al. Demographic and clinical features of neuromyelitis optica: A review. Mult Scler. 2015;21:845–53.
Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology. 2006;66:1485–9.
Tillema JM, McKeon A. The spectrum of neuromyelitis optica (NMO) in childhood. J Child Neurol. 2012;27:1437–47.
Matiello M, Lennon VA, Jacob A, et al. NMO-IgG predicts the outcome of recurrent optic neuritis. Neurology. 2008;70:2197–200.
Weinshenker BG, Wingerchuk DM, Vukusic S, et al. Neuromyelitis optica IgG predicts relapse after longitudinally extensive transverse myelitis. Ann Neurol. 2006;59:566–9.
Lucchinetti CF, Mandler RN, McGavern D, et al. A role for humoral mechanisms in the pathogenesis of Devic’s neuromyelitis optica. Brain. 2002;125:1450–61.
Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202:473–7.
Saadoun S, Waters P, Bell BA, Vincent A, Verkman AS, Papadopoulos MC. Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain. 2010;133:349–61.
Jarius S, Wildemann B. AQP4 antibodies in neuromyelitis optica: diagnostic and pathogenetic relevance. Nat Rev Neurol. 2010;6:383–92.
Bradl M, Misu T, Takahashi T, et al. Neuromyelitis optica: Pathogenicity of patient immunoglobulin in vivo. Ann Neurol. 2009;66:630–43.
Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol. 2007;6:805–15.
Misu T, Fujihara K, Nakashima I, Sato S, Itoyama Y. Intractable hiccup and nausea with periaqueductal lesions in neuromyelitis optica. Neurology. 2005;65:1479–82.
Iorio R, Lucchinetti CF, Lennon VA, et al. Syndrome of inappropriate antidiuresis may herald or accompany neuromyelitis optica. Neurology. 2011;77:1644–6.
Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85:177–89.
Sepúlveda M, Armangue T, Sola-Valls N, et al. Neuromyelitis optica spectrum disorders: comparative study according to the phenotype and serostatus. Neurol Neuroimmunol Neuroinflamm. (in press).
Höftberger R, Sepulveda M, Armangue T, et al. Antibodies to MOG and AQP4 in adults with neuromyelitis optica and suspected limited forms of the disease. Mult Scler. 2015;21:866–74.
Jiao Y, Fryer JP, Lennon VA, et al. Updated estimate of AQP4-IgG serostatus and disability outcome in neuromyelitis optica. Neurology. 2013;81:1197–204.
Zamvil SS, Slavin AJ. Does MOG Ig-positive AQP4-seronegative opticospinal inflammatory disease justify a diagnosis of NMO spectrum disorder? Neurol Neuroimmunol Neuroinflamm. 2015;2:e62.
Pittock S, Lucchinetti C. Neuromyelitis optica and the evolving spectrum of autoimmune aquaporin-4 channelopathies: a decade later. Ann N Y Acad Sci. 2015;doi: 10.1111/nyas.12794.
Guo Y, Lennon VA, Popescu BF, et al. Autoimmune aquaporin-4 myopathy in neuromyelitis optica spectrum. JAMA Neurol. 2014;71:1025–9.
Verkman AS, Phuan PW, Asavapanumas N, Tradtrantip L. Biology of AQP4 and anti-AQP4 antibody: Therapeutic implications for NMO. Brain Pathol. 2013;23:684–95.
Waters PJ, McKeon A, Leite MI, et al. Serologic diagnosis of NMO: A multicenter comparison of aquaporin-4-IgG assays. Neurology. 2012;78:665–71.
Mckeon A, Lennon VA, Lotze T, et al. CNS aquaporin-4 autoimmunity in children. Neurology. 2008;71:93–100.
Banwell B, Tenembaum S, Lennon VA, et al. Neuromyelitis optica-IgG in childhood inflammatory demyelinating CNS disorders. Neurology. 2008;70:344–52.
Hacohen Y, Absoud M, Woodhall M, et al. Autoantibody biomarkers in childhood-acquired demyelinating syndromes: results from a national surveillance cohort. J Neurol Neurosurg Psychiatry. 2014;85:456–61.
Lotze TE, Northrop JL, Hutton GJ, Ross B, Schiffman JS, Hunter JV. Spectrum of pediatric neuromyelitis optica. Pediatrics. 2008;122:e1039–47.
Collongues N, Marignier R, Zéphir H, et al. Long-term follow-up of neuromyelitis optica with a pediatric onset. Neurology. 2010;75:1084–8.
Wang KC, Lin KH, Lee TC, et al. Poor responses to interferon-beta treatment in patients with neuromyelitis optica and multiple sclerosis with long spinal cord lesions. PLoS One. 2014;9:e98192.
Min JH, Kim BJ, Lee KH. Development of extensive brain lesions following fingolimod (FTY720) treatment in a patient with neuromyelitis optica spectrum disorder. Mult Scler. 2012;18:113–5.
Kim SH, Jeong IH, Hyun JW, et al. Treatment outcomes with rituximab in 100 patients with neuromyelitis optica: Influence of FCGR3A polymorphisms on the therapeutic response to Rituximab. JAMA Neurol. 2015;72:989–95.
Pittock SJ, Lennon VA, McKeon A, et al. Eculizumab in AQP4-IgG-positive relapsing neuromyelitis optica spectrum disorders: An open-label pilot study. Lancet Neurol. 2013;12:554–62.
Araki M, Matsuoka T, Miyamoto K, et al. Efficacy of the anti-IL-6 receptor antibody tocilizumab in neuromyelitis optica: a pilot study. Neurology. 2014;82:1302–6.
Hammer O. CD19 as an attractive target for antibody-based therapy. MAbs. 2012;4:571–7.
Tradtrantip L, Zhang H, Saadoun S, et al. Anti-aquaporin-4 monoclonal antibody blocker therapy for neuromyelitis optica. Ann Neurol. 2012;71:314–22.
Mader S, Gredler V, Schanda K, et al. Complement activating antibodies to myelin oligodendrocyte glycoprotein in neuromyelitis optica and related disorders. J Neuroinflammation. 2011;8:184.
Baumann M, Sahin K, Lechner C, et al. Clinical and neuroradiological differences of paediatric acute disseminating encephalomyelitis with and without antibodies to the myelin oligodendrocyte glycoprotein. J Neurol Neurosurg Psychiatry. 2014;86:265–72.
Probstel AK, Dornmair K, Bittner R, et al. Antibodies to MOG are transient in childhood acute disseminated encephalomyelitis. Neurology. 2011;77:580–8.
Di Pauli F, Mader S, Rostasy K, et al. Temporal dynamics of anti-MOG antibodies in CNS demyelinating diseases. Clin Immunol. 2011;138:247–54.
Hacohen Y, Absoud M, Deiva K, et al. Myelin oligodendrocyte glycoprotein antibodies are associated with a non-MS course in children. Neurol Neuroimmunol Neuroinflamm. 2015;2:e81.
Rostásy K, Mader S, Hennes EM, et al. Persisting myelin oligodendrocyte glycoprotein antibodies in aquaporin-4 antibody negative pediatric neuromyelitis optica. Mult Scler. 2013;19:1052–9.
Kitley J, Woodhall M, Waters P, et al. Myelin-oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotype. Neurology. 2012;79:1273–7.
Rostasy K, Mader S, Schanda K, et al. Anti–myelin oligodendrocyte glycoprotein antibodies in pediatric patients with optic neuritis. Arch Neurol. 2012;69:752–6.
Huppke P, Rostasy K, Karenfort M, et al. Acute disseminated encephalomyelitis followed by recurrent or monophasic optic neuritis in pediatric patients. Mult Scler. 2013;19:941–6.
Fernandez-Carbonell C, Vargas-Lowy D, Musallam A, et al. Clinical and MRI phenotype of children with MOG antibodies. Mult Scler. 2016;22:174–84.
Kitley J, Waters P, Woodhall M, et al. Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies: A comparative study. JAMA Neurol. 2014;71:1–8.
Sato DK, Callegaro D, Lana-Peixoto MA, et al. Distinction between MOG antibodypositive and AQP4 antibody-positive NMO spectrum disorders. Neurology. 2014;82:474–81.
Ikeda K, Kiyota N, Kuroda H, et al. Severe demyelination but no astrocytopathy in clinically definite neuromyelitis optica with anti-myelin-oligodendrocyte glycoprotein antibody. Mult Scler. 2015;21:656–9.
Srivastava R, Aslam M, Kalluri SR, et al. Potassium channel KIR4.1 as an immune target in multiple sclerosis. N Engl J Med. 2012;367:115–23.
Kraus V, Srivastava R, Kalluri SR, et al. Potassium channel KIR4.1-specific antibodies in children with acquired demyelinating CNS disease. Neurology. 2014;82:470–3.
Brickshawana A, Hinson A, Romero M, et al. Investigation of the KIR4.1 potassium channel as a putative antigen in patients with multiple sclerosis: a comparative study. Lancet Neurol. 2014;13:795–806.
Nerrant E, Salsac C, Charif M, et al. Lack of confirmation of anti-inward rectifying potassium channel 4.1 antibodies as reliable markers of multiple sclerosis. Mult Scler. 2014;20:1699–703.
Brill L, Goldberg L, Karni A, et al. Increased anti-KIR4.1 antibodies in multiple sclerosis: could it be a marker of disease relapse? Mult Scler. 2015;21:572–9.
Filippi M, Rocca MA, Lassmann H. KIR4.1: another misleading expectation in multiple sclerosis? Lancet Neurol. 2014;13:753–5.
Armangue T, Leypoldt F, Dalmau J. Autoimmune encephalitis as differential diagnosis of infectious encephalitis. Curr Opin Neurol. 2014;27:361–8.
Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol. 2011;10:63–74.
Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: An observational cohort study. Lancet Neurol. 2013;12:157–65.
Armangue T, Titulaer MJ, Málaga I, et al. Pediatric anti-N-methyl-D-aspartate receptor encephalitis-clinical analysis and novel findings in a series of 20 patients. J Pediatr. 2013;162:850–6. e2.
Dalmau J, Gleichman AJ, Hughes EG, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008;7:1091–8.
Ramberger M, Bsteh G, Schanda K, et al. NMDA receptor antibodies: A rare association in inflammatory demyelinating diseases. Neurol Neuroimmunol Neuroinflamm. 2015;2:e141.
Gresa-Arribas N, Titulaer MJ, Torrents A, et al. Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol. 2014;13:167–77.
Carvajal-González A, Leite MI, Waters P, et al. Glycine receptor antibodies in PERM and related syndromes: Characteristics, clinical features and outcomes. Brain. 2014;137:2178–92.
Ariño H, Gresa-Arribas N, Blanco Y, et al. Cerebellar ataxia and glutamic acid decarboxylase antibodies: Immunologic profile and long-term effect of immunotherapy. JAMA Neurol. 2014;71:1009–16.
Martinez-Hernandez E, Sepulveda M, Rostásy K, et al. Antibodies to aquaporin 4, myelin-oligodendrocyte glycoprotein, and the glycine receptor α1 subunit in patients with isolated optic neuritis. JAMA Neurol. 2015;72:187–93.
Acknowledgements
We thank Myrna Rosenfeld for critical review of the manuscript and all participating physicians and patients for their generous contribution to this research. This work was supported in part by Instituto Carlos III, Madrid, Spain (CM14/00081 TA); Dodot Procter & Gamble research grant sponsored by Asociación Española de Pediatría (AEP) (DN040579, TA); and Fundació Marató TV3 (20141830, TA, MS, FG, AS).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Dr. Armangue has full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: Armangue, Saiz. Drafting of the manuscript: Armangue, Yeshokumar, Saiz. Critical revision of the manuscript for important intellectual content: All authors approved the final version of the manuscript.
Authors’ information
Dr. Thaís Armangue received her MD degree with honors from the Autonomous University of Barcelona School of Medicine in 2006 and her PhD cum laude from the University of Barcelona in 2015. She did residency training in Pediatrics (2007–2011), a fellowship in Pediatric Neurology (2011–2013) at Vall d’Hebron Hospital in Barcelona, Autonomous University of Barcelona, and a clinical-research fellowship in Neuroimmunology (2012–2015) at Institut d’Investigacions Biomèdiques August Pi I Sunyer (IDIBAPS)-Hospital Clinic, University of Barcelona, Barcelona, Spain. Currently she is a member of the Neuroimmunology Program at IDIBAPS-Hospital Clinic (Barcelona, Spain) and coordinates the Pediatric Neuroimmunology Unit at Sant Joan de Déu Childrens Hospital (University of Barcelona) in Barcelona. Her research involves both clinical and basic science studies to identify new antigens associated with pediatric autoimmune encephalitis and demyelinating disorders. Dr. Armangue is a member of the International Pediatric Multiple Sclerosis Study Group (IPMSSG).
Dr. Anusha K. Yeshokumar received her undergraduate degree in Neuroscience at Johns Hopkins University in 2006 and her MD degree from Jefferson Medical College in 2011. She did residency training in Pediatrics (2011–2013) at the Maria Fareri Children's Hospital in New York and is currently completing residency training in Pediatric Neurology at Johns Hopkins. In 2016 she will enter the fellowship program in Pediatric Demyelinating and Inflammatory Brain Diseases at the Children’s Hospital of Philadelphia (CHOP). Dr. Yeshokumar’s research centers on understanding the clinical characteristics of pediatric autoimmune encephalitis and demyelinating disorders and the effect of these disorders on brain development and cognitive function.
Dr. Maria Sepúlveda received her MD from the University of Barcelona in 2005 and completed a residency program in Neurology at the Hospital del Mar in Barcelona in 2010. Since 2010 she has been a member of Neuroimmunology and Multiple Sclerosis Unit at Hospital Clinic, Barcelona. Dr. Sepulveda’s research is focused on the identification of serological and clinical markers of multiple sclerosis and neuromyelitis optica spectrum disorders.
Professor Francesc Graus received his MD and PhD from the University of Barcelona. He is chair of the Neurology Department of Hospital Clinic (Barcelona), and director of the Clinical and Experimental Neuroimmunology Laboratory Program at IDIBAPS (Barcelona). Dr. Graus’s research is focused in the development of clinical and biological markers of paraneoplastic disorders and autoimmune encephalitis.
Dr. Albert Saiz received his MD and PhD from the University of Barcelona. He is head of the Neuroimmunology-Multiple Sclerosis Unit of Hospital Clinic (Barcelona), and a researcher in the Neuroimmunology Program at IDIBAPS. Dr. Saiz’s research is focused in the development of clinical, biological and imaging markers of demyelinating disorders and the development of new therapeutics strategies.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Armangue, T., Yeshokumar, A.K., Sepúlveda, M. et al. Antibodies in acquired demyelinating disorders in children. Mult Scler Demyelinating Disord 1, 5 (2016). https://doi.org/10.1186/s40893-016-0008-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40893-016-0008-9