
Abstract
Background
Moderate to severe anxiety disorders such as obsessive-compulsive disorder (OCD), post-traumatic stress disorder (PTSD), social phobia and panic disorder are common, and affect approximately 11–16% of women in pregnancy. Psychological treatments for anxiety disorders, primarily cognitive behaviour therapy (CBT), have a substantial evidence base and recently time-intensive versions have been found as effective as weekly treatments. However, this has not been trialled in women who are pregnant, where a shorter intervention may be desirable.
Methods
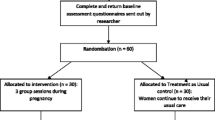
The ADEPT study is a feasibility randomised controlled trial with two parallel intervention groups. Time-intensive one-to-one CBT and standard weekly one-to-one CBT delivered during pregnancy will be compared. Feasibility outcomes including participation and follow-up rates will be assessed, alongside the acceptability of the interventions using qualitative methods.
Discussion
The study will provide preliminary data to inform the design of a full-scale randomised controlled trial of a time-intensive intervention for anxiety during pregnancy. This will include information on the acceptability of time-intensive interventions for pregnant women with anxiety disorders.
Trial registration
https://doi.org/10.1186/ISRCTN81203286 prospectively registered 27/6/2019.
Similar content being viewed by others

Background
The mental health of pregnant women and those with a baby up to 1 year (known as the perinatal period) is a priority due to the potential impact on both mother and child [1]. Anxiety disorders are common and functionally impairing, affecting 11–16% of women in pregnancy and 8–17% of women in the postpartum period [2–5]. Pregnancy can exacerbate or trigger anxiety disorders and can also elicit new fears related to pregnancy and parenting [6]. For most women, antenatal anxiety disorders persist into the postpartum period [4, 7], and they can increase the risk of postpartum depression [8]. Postpartum anxiety disorders have been associated with impaired maternal functioning [9–12], excessive infant crying and feeding problems including lower rates of breastfeeding. Children of perinatally anxious mothers are at raised risk of experiencing emotional and behavioural problems [13–16].
It is therefore important to identify and treat antenatal anxiety disorders quickly, to ameliorate or prevent these short- and long-term outcomes for both mother and child. Shorter exposure of the developing fetus to the elevated levels of maternal cortisol associated with anxiety may protect the child whilst successful antenatal treatment would also improve quality of life for the mother and potentially reduce the impact on parenting. The need for timely treatment is reflected in clinical practice guidelines [17]. However, the National Institute for Health and Care Excellence (NICE) guidelines for perinatal mental health [17] highlight the need for more research on treating moderate to severe anxiety disorders, in particular obsessive-compulsive disorder (OCD), panic, post-traumatic stress disorder (PTSD) and social phobia (recommendation 7.7.2.4). Each of these disorders can be triggered or exacerbated in the context of pregnancy and can affect mother-infant interactions and parenting [18].
Pregnant women prefer psychological treatments to pharmacotherapy, but there are barriers to such interventions including having time to attend sessions [19]. UK primary mental health care sites have reported dropout rates prior to treatment for perinatal women of up to 40% [20]. Shorter, but more intensive treatment could therefore be an innovative and welcome format that may improve engagement and optimise outcomes for both mother and baby.
Psychological treatments for anxiety disorders, primarily cognitive behaviour therapy (CBT), have a substantial evidence base [21–23]. Women are usually offered one-to-one disorder specific CBT based on best available evidence [24]. Individual CBT is ‘semi-idiographic’, that is, based on shared principles but tailored to the individual context and needs of the client. Modifications may be made according to the client’s physical state, such as not running upstairs in a panic exposure exercise if the person is unable, instead identifying other means to test relevant beliefs. Such modifications in pregnancy have been suggested and there is some evidence for the use of exposure-based CBT during pregnancy.
CBT is usually delivered in approximately 12-h-long weekly sessions, depending on the presenting disorder. Time-intensive CBT treatments (IN-CBT) have been trialled in (non-perinatal) patients with OCD, PTSD, social phobia and panic disorder with equivalent outcomes to standard weekly treatments, achieved in a much shorter time frame of 1 to 2 weeks [25–28]. This format has been found acceptable to patients. IN-CBT for postpartum OCD has been found to be safe and effective in the reduction of maternal OCD symptoms and acceptable to mothers [29, 30]. Given the clear aim of delivering fast and effective treatment, ideally before the baby is born, IN-CBT could be a helpful modification to traditional weekly CBT for pregnant women with anxiety disorders and may be more beneficial in terms of longer-term outcomes. Specific evidence is therefore needed to establish whether IN-CBT approaches are acceptable, produce equivalent engagement and adherence with treatment during pregnancy, are efficacious during pregnancy and helpful for maternal symptoms and parenting in the postpartum.
Study aims and objectives
The primary objective of this research is to assess the feasibility of a definitive trial of antenatal IN-CBT compared with standard weekly antenatal CBT (treatment as usual) for women experiencing OCD, PTSD, social phobia or panic disorder in pregnancy. These disorders have evidence for the use of intensive approaches. Outcomes will assess the feasibility and acceptability of recruitment methods; recruitment rates and participants’ willingness to be randomised; the acceptability of assessment measures, intervention mode and intervention delivery; treatment fidelity; follow-up rates; and estimates of sample size parameters. We will use this information to inform a full-scale randomised controlled trial (RCT).
The secondary objectives are to collect and summarise clinical outcomes and physical and mental health service use for health economic evaluation.
Methods
Study design
This study is a feasibility 1:1 randomised controlled trial with two parallel intervention groups. Methods are presented as per the Standard Protocol Items: Recommendations for International Trials (SPIRIT) [31]. All elements of the SPIRIT checklist are reported below.
Participants, interventions and outcomes
Study setting
The study is set within adult mental healthcare services in English National Health Service (NHS) settings. As the national publicly funded healthcare system for England, the NHS provides healthcare for all legal residents in the UK, and mental healthcare is amongst the services free at the point of use.
The study setting will be four outpatient Improving Access to Psychological Therapies (IAPT) services within South London and Maudsley (SLaM) NHS Foundation Trust in South East London. The four outpatient IAPT services (Lambeth, Lewisham, Southwark and Croydon) have a named perinatal lead or experienced therapist who will provide standard weekly and IN-CBT treatments within the trial. The Centre for Anxiety Disorders and Trauma (CADAT) is part of Lambeth, Lewisham and Southwark IAPT services and will provide both treatments.
Participant recruitment and eligibility criteria
Women will be recruited to the trial in one of 2 ways:
-
a)
Women attending antenatal booking clinics in a South East London maternity service (in the boroughs of Lambeth, Lewisham, Southwark or Croydon only) may be approached by their midwife with information about the study, who may contact the researchers on their behalf if the woman is interested and agrees. Alternatively, women may self-refer to the trial if they feel they are experiencing symptoms of anxiety consistent with one of the anxiety disorders being treated. Advertisements (e.g. leaflets and posters) for self-referral to the trial will be positioned in maternity service waiting rooms and GP surgeries and other settings for local pregnant women, e.g. children’s centres to encourage self-referral.
-
b)
Women may be referred to the trial by a therapist in the IAPT services if the therapist assessed the woman as having an anxiety disorder and potentially suitable for the trial. Clinicians will be regularly reminded about the study and flyers and posters made available.
Inclusion criteria
Women must have the following characteristics prior to randomisation:
-
1.
Aged ≥ 18 years
-
2.
Pregnant, between 12 and 25 weeks gestation
-
3.
Meet criteria for Diagnostic and Statistical Manual of Mental Disorders Fifth Edition (DSM-V) for OCD, PTSD, social anxiety or panic disorder on the Structured Clinical Interview DSM-V (SCID-V)
-
4.
Eligible for referral to Lambeth, Lewisham, Southwark or Croydon IAPT services (i.e. has local general practitioner, common mental disorder and level of risk manageable within primary care)
-
5.
Available (by self-report) for either IN-CBT or standard weekly treatment
-
6.
Not taking psychotropic medication or taking stable dose of medication for at least 6 weeks
-
7.
Able to provide informed consent
Exclusion criteria
The exclusion criteria are as follows:
-
1.
Pregnant women with a primary DSM-V depressive disorder, affective or psychotic disorder or current problems with substance abuse
-
2.
Pregnant women with ‘complex PTSD’ (defined as prolonged multiple traumas affecting a number of domains)
-
3.
Pregnant women who have a medically high-risk pregnancy at the time of recruitment involving significant additional management (e.g. multiple sclerosis, lupus, polycystic ovary syndrome)
-
4.
Pregnant women who are receiving cognitive behaviour therapy or another individual or group psychological therapy
-
5.
Pregnant women who are unable to read English adequately to complete questionnaires
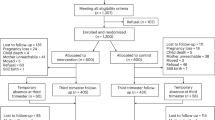
The research psychologist will obtain informed consent from potential trial participants who will send a signed consent form. Women will be given at least 24 h after receiving the information sheet to decide whether to proceed with consent and baseline measures. Figure 1 shows the participant timeline through the study.

Schematic representation of time in the study
Assignment of interventions
Sequence generation
Participants will be randomly allocated using a 1:1 ratio to (1) time-intensive CBT (IN-CBT) or (2) standard weekly CBT (i.e. treatment as usual). Participants in either allocation group will receive treatment delivered by a high-intensity cognitive behaviour therapist (HI CBT therapist). Randomisation will be conducted via an independent online system based at the King’s Clinical Trials Unit (King’s CTU) based at King’s College London. Random allocation will be at the level of the individual participant and minimisation will be used to achieve balance amongst minimisation factors; disorder (OCD, panic, PTSD, social phobia) and site (Croydon, Lambeth, Lewisham, Southwark).
Concealment mechanism
Participants will be randomised to the trial groups (1:1 allocation) to ensure allocation concealment is high. Any preference on allocation will be recorded prior to randomisation.
Implementation
The allocation sequence is generated by the KCTU online system. The research psychologist will enrol participants. Once an eligible woman has given consent to participate in the study and completed baseline measures, the research psychologist will use the online system to perform the randomisation. The research psychologist will communicate information about the allocation group to the participant (either time-intensive CBT treatment (IN-CBT) or standard weekly CBT treatment) and to the therapist who will arrange an appointment with the participant at the earliest mutually convenient opportunity.
Blinding
For each allocation to the IN-CBT or standard weekly treatment group, the participant, their therapist and the research psychologist will be aware of the allocation. The research assistant will be blind to the group allocation and will not have access to allocation databases. The research assistant will collect outcome measures at baseline, 1 month and 3 months (primary outcome point) and will be blind. They will conduct the qualitative interview last as it will likely lead to unblinding. The senior statistician will be blind to the group allocation until the end of the trial.
The senior statistician will become unblind once the primary and secondary analyses have been performed. The Statistical Analysis Plan will be drafted by the trial statistician and the senior statistician will contribute fully blinded. The outcome assessor will be unblinded at the end of final outcome assessments by conducting the qualitative interview, as the study is not double blind. This occurs after the other assessment information has been collected.
Data collection and management
Baseline assessment
Baseline measures will be collected by the research psychologist or research assistant prior to the assignment of participants to the intervention groups. Baseline measures will be collected via weblink using existing data collection methods in IAPT services or hard copies as preferred by participants and will be completed in participants own time. Initial clinical interviews will be conducted over the phone at a suitable time for the mother, or in person if she prefers. As per standard practice in IAPT services, anxiety, depression and disorder specific measures will be collected at each point of contact. Participants will be asked to complete the additional measures at the baseline and outcome points. The postnatal follow-up video and qualitative interview (see below for details) will be collected during a visit to the participant’s home by the research assistant who will be blind to the allocation status of the participant. Assessments may take place via phone or video link using Microsoft Teams, depending on current restrictions due to the Coronavirus.
A list of measures collected during the study and the timepoints of data collection are presented in Table 1.
Measures related to primary objectives
The primary feasibility outcomes will be determined through analysis of the recruitment and follow up data. Therapist time, including participant contact and assessment of time spent in other indirect participant related activities (e.g. supervision, training, administration etc.), will be recorded by therapists using standardised clinical notes. Therapy content and adherence will be determined by analysis of these notes.
As is standard practice in CBT, therapy sessions will be audio-recorded by therapists. A random selection of therapy tapes from each therapist will be rated by assessors blind to the aims of the study using a standardised measure of cognitive therapy skills, the Cognitive Therapy Rating Scale (CTS-R [32];).
Measures related to secondary objectives
Initial clinician interview collected at baseline
Structured clinical interview for DSM-V (SCID-V [33];). This semi-structured interview is used by clinicians to establish DSM-V diagnoses, which will be part of initial screening for inclusion in the trial [34].
Routinely collected IAPT data collected at baseline, each clinical session and 3 months postpartum:
-
1.
Demographic (at baseline only)—maternal age, ethnicity, occupation, relationship status, whether they are taking psychotropic medication and pregnancy stage will be recorded.
-
2.
*GAD-7 [35] is a well validated and widely used 7 item measure of anxiety and a key IAPT outcome measure used routinely. A change of 4 or more on the GAD has been found to be clinically significant across anxiety disorders [36]. *This is the potential primary outcome measure for a future evaluation trial.
-
3.
PHQ-9 [37] is a well-validated 9-item questionnaire used to measure symptoms of depression. It is also a key IAPT outcome measure.
-
4.
The Work and Social Adjustment Scale [38] is a 5-item patient self-report measure, which assesses the impact of a person’s mental health difficulties on their ability to function in terms of work, home management, social leisure, private leisure and personal or family relationships.
-
5.
Disorder specific measures: one of the following will be used depending on primary disorder.
-
(a)
OCD: Obsessive Compulsive Inventory-Revised (OCI [39];). This is a 42-item self-report inventory concerning symptoms of OCD. The internal consistency for the full scale is high (0.86–0.95). The OCI also shows good discriminative validity and is reliable to measure change in symptoms over time.
-
(b)
Panic: Mobility Inventory (alone) [40]. This self-report scale requires respondents to rate avoidance of a range of specific situations over the last week on a scale of 0–4. The inventory has good reliability, validity and sensitivity to change.
-
(c)
PTSD: Impact of Events Scale (IES [41]). This is a 22 item self-report scale of symptoms of PTSD. The scale has excellent internal consistency [42].
-
(d)
Social Phobia: The Social Phobia Inventory (SPIN [43]) has 17 items and a cut-off score of 19 or above. For each item, respondents rate from 0 to 4 how bothered they have been by the item during the past week. The total score provides a measure of the severity of social phobia and the item has good sensitivity to change.
Trial-specific measures
Working alliance inventory–Short Revised–collected after 2 h of treatment
-
6.
Working Alliance Inventory–Short Revised [44]. This 12-item questionnaire will be completed by therapists and participants in both arms.
Pregnancy Anxiety–collected at start of treatment and late pregnancy follow-up session
-
7.
Pregnancy-Related Anxiety Questionnaire (PRAQ [45];). This 10-item self-report questionnaire assesses anxiety related to childbirth and will be administered at baseline and the last antenatal appointment treatment session. The pregnancy-related anxiety scale has acceptable internal reliability (Cronbach’s α = .78).
Infancy related measures—collected at 3 months postpartum
-
8.
Postpartum Bonding Questionnaire (PBQ) [46]. This self-report instrument consists of 25 items to be rated on a scale of 0 (‘never’) to 5 (‘always’) and assesses the maternal perception of her felt bonding with the infant.
-
9.
Mother infant interactions
Mother-infant interactions at 3 months postpartum will be captured in a 3-min video taken during play and nappy change at home and subsequently assessed by a trained rater using the CARE Index [47]. This tool has been widely used in scientific research with mothers that have mental health difficulties as early as 1 to 5 months postpartum [48–50] and validated for use with families from different social classes and cultural backgrounds [51]. Coding comprises seven aspects of adult and infant dyadic behaviour: four aspects concentrate on affect (facial expression, verbal expression, affection and body contact) and three focus on temporal contingencies (turn-taking, control and developmental appropriateness of chosen activity). Each aspect of adult and infant behaviour is evaluated individually and summed to make seven scale scores. For adults, these are sensitivity, unresponsiveness and controlling. Infants (birth to 15 months of age) are coded on cooperativeness, difficultness, compulsivity and passivity. Interactions receive a score on each aspect of adult and infant behaviour and scores are then summed to create the seven scale scores, each on a range from 0 to 14 [47]. For example, a ‘sensitive dyad’, the mother must achieve a score of 11 or higher on the sensitivity scale. A score of 7 or more is required to rate the interaction as ‘adequate’. Five to 6 points mark the ‘inept’ range and suggest the need for parental education. Four or fewer points are considered as in the ‘high risk’ range, implying risk of abuse or neglect. Coding of the interaction takes between approximately 30–40 min. Video-taped interactions will be coded by trained, reliable coders, as recommended by Crittenden [47]. To reduce measurement bias, the coders will be unaware of the hypotheses that are being tested in the study and blind to the mental health status of the women.
Health economic measures collected at 3 months.
-
10.
Adult Service Use Measure (ADSUS, unpublished [52]). The AD-SUS is a measure of resource use developed for use in mental health populations and a version adapted for use in prenatal populations as part of an NIHR PGfAR will be used (the ESMI study). This measure will be used to capture maternal and child utilization of all health and social care services (including health visitor, GP visits).
Measures 8–10 have been included to assess the potential impact on parenting and service use.
Qualitative data
Qualitative interviews will be conducted with an estimated 15–20 women undergoing IN-CBT and 15–20 women undergoing standard CBT, or until inductive thematic saturation is reached, and new data is easily accommodated within the thematic framework to further examine the acceptability and feasibility of the intervention and study design [53]. Interviews will explore experiences of having an anxiety disorder in pregnancy, experiences of treatment including perceived benefits and limitations and challenges during treatment and in postpartum adjustment. Experiences of recruitment, randomisation and assessments will also be examined. Topics will be derived in collaboration with the project advisory group. Purposive sampling will be conducted on the basis of diagnosis, socio-economic status, ethnicity, recruitment source and session attendance to explore a range of perspectives. Data collection and analysis will be conducted in parallel with the topic guide and sampling strategy amended iteratively to explore the diversity of data. Interviews will take place at the final outcome point 3 months postpartum. These will be recorded and transcribed. All therapists will also be interviewed to examine how IN-CBT and standard CBT were delivered in practice (e.g. treatment fidelity, ease of delivery, perceived engagement).
Details of interventions
Cognitive behaviour therapy interventions are the NICE guideline recommended treatment for anxiety disorders and are considered effective [21–23]. Psychological interventions are generally preferred by pregnant women and this study seeks to investigate a time-intensive approach given the time parameter of pregnancy. Therefore time-intensive and standard weekly CBT approaches will be compared.
There are well developed specific protocols and techniques for using CBT to treat anxiety disorders [54]. In general, CBT follows the course of establishing the participant’s goals for therapy and developing a patient-specific formulation based on the relevant model (e.g. Salkovskis for OCD; Wells & Clark for Social Phobia; Ehlers & Clark for PTSD; Clark for Panic disorder). The formulation identifies relevant beliefs and behaviours that maintain the anxiety disorder. Cognitive and behavioural strategies are then used to help patients change the beliefs and behaviours that maintain their anxiety disorder. A standard modification to treatment in the perinatal context would be to include planning and support for the demands of the postpartum environment whilst the person is attending antenatal treatment. All participants and therapists audio-record sessions (as per standard clinical practice) to listen to between appointments and participants will be encouraged to apply new learning and strategies as homework. The content of treatment will be the same for both intervention arms.
Time-intensive CBT
This will comprise 8–10 h (depending on the disorder) of one-to-one CBT delivered in 4–5 sessions over 1–2 weeks, delivered at the earliest convenient point between 12 and 36 weeks of pregnancy. The CBT treatment will be delivered by a high-intensity cognitive behaviour (HI CBT) therapist, which is standard practice within IAPT services. Two follow up sessions of 1 h each will then be offered by the HI CBT therapist which will include one in late pregnancy (33–35 weeks’ gestation) and one at 1m postpartum.
Standard weekly CBT (treatment as usual)
Standard weekly CBT will comprise 8–10 hours (depending on the disorder) of one to one CBT on a one hour per week basis. This is termed ‘high intensity’ CBT in IAPT services and will be offered to all women who participate in the trial and are randomised to standard weekly CBT. The CBT treatment will be delivered by a High Intensity Cognitive Behaviour (HI CBT) Therapist, which is standard practice within IAPT services. Two follow up sessions of one hour will then be offered by the HI CBT Therapist which will include one in late pregnancy (33–35 weeks’ gestation), one at 1m postpartum.
Treatments may take place via phone or video link using Microsoft Teams, depending on current restrictions due to the Coronavirus.
Provisions for post-trial care
Women requiring and wishing for further intervention after the end of the trial will be referred or signposted to an appropriate intervention. This may be for further psychological therapy within IAPT or within more specialised perinatal mental health services.
Criteria for discontinuing or modifying allocated interventions
Adverse events will be monitored and recorded including the mental and physical health of the mother and any pregnancy-related events. Should participants deteriorate clinically in terms of significantly increased symptoms of anxiety or depression, or an increase in risk to themselves or others, then the intervention will be stopped and a referral made to specialist perinatal services and/or other services as clinically appropriate.
Strategies to improve adherence to interventions
Fidelity will be defined as delivering all key treatment components during treatment and will be established by a checklist of key components of treatment derived from core competencies for specific disorders using treatment notes [23]. This will be completed by the therapist at the end of each session. Standardised therapy note sheets will be kept and checked to establish fidelity against the checklist. Therapists will also be required to submit a random selection of therapy recordings to be rated for fidelity to CBT on an established scale, the Cognitive Therapy Rating Scale (CTS-R).
Client engagement and adherence to therapy will be rated by therapist at each session on a 4-point Likert scale, number of sessions attended will be recorded and the participants’ perspective will be assessed in detail in a qualitative interview at 3 months.
Outcome progression criteria
Primary feasibility outcomes:
-
(i)
if it is possible to identify/recruit patients via (a) information provided in healthcare settings at booking visits/by midwives/self-referral and (b) approach by a primary care therapist. We require a minimum of 30% of those approached to be eligible participants from each recruitment method for each to be deemed feasible. An acceptable recruitment rate (number of consented participants) would be at least 3 participants/month
-
(ii)
If participants are willing to be randomised. We require that 70% of eligible participants are randomised to be deemed feasible
-
(iii)
If the intervention is received as intended in both arms using a fidelity content checklist. A minimum of 70% of participants in the trial would need to complete > 60% of each intervention in hours for each to be deemed feasible, i.e. 7.2 h out of 12 [28]. For completers in the intensive arm, these treatment hours will need to be completed in the 2-week window
-
(iv)
Acceptability of both interventions to participants will be determined by qualitative investigation enquiring about experiences and positive and any negative effects of all aspects of treatment, and a 3-point rating scale to assess how useful the treatment was for anxiety and parenting
-
(v)
Establishing the parameters needed in order to estimate the sample size for a full trial. For example, standard deviation of the potential primary outcome measure GAD-7
-
(vi)
Participation and data completion at 3m outcome assessment; a follow up rate of > 70% is required to determine feasibility
-
(vii)
The acceptability of assessment measures to participants; this will be determined by qualitative interview and brief 3-point rating scales asking if it was acceptable and clear
Secondary clinical outcome measures:
-
(i)
Clinical outcome measures for depression, anxiety, overall functioning and anxiety disorder symptoms collected at 3 months postpartum
-
(ii)
Health service use at 3 months postpartum
Plans to promote participant retention and complete follow-up
Participants will receive an incentive (£10) for the additional burden incurred from completing assessments at two time points. Participants will receive £10 for participating in the baseline assessment and £10 for participating in final outcome point assessments. They will be encouraged to take part in the final outcome point assessment regardless of treatment completion.
Sample size estimation
In line with guidance on feasibility studies, no power calculation has been carried out [55]. A sample size of 30 in each intervention group has been recommended to answer questions relating to feasibility [56, 57].
Data management
The trial statistician will assist with management of the trial data which will be split into 3 databases. The participant main database and therapy database will be stored in separate SPSS files on a network drive only accessible by the CI and trial statistician. The AE (adverse events) database will be stored as a password-protected Excel file on a network drive accessible by the CI only. All databases will be backed up periodically (approx. every 3 months), by creating a date-stamped ZIP file storing all databases and storing in a subfolder titled ‘Archives’. This will be carried out by both the trial statistician and the CI separately.
All research data will be pseudonymised using unique identification numbers and stored without contact details (names or addresses). Associations between participants' contact details and identification numbers will be stored in a separate encrypted electronic password-protected database. Access to this document will be restricted to the Chief Investigator. All data will be held on a secure database on an encrypted, password-protected computer and access to it will be restricted to the research team. Audio files of the qualitative interviews will be retained until they have been transcribed to written form. Transcriptions of qualitative interviews will be completed as soon as possible after collection, anonymised and uploaded to the computer software programme, QSR N-VIVO. Hardcopies of study consent forms held by the central research team will be kept in a locked cabinet at the Institute of Psychiatry, Psychology & Neuroscience and retained for 7 years post research data analysis.
Questionnaire data
Questionnaire data will be collected via weblink using existing data collection methods in IAPT services and/or hard copies as preferred, completed in participants own time. Initial clinician interviews will be conducted over the phone at a suitable time for the mother, or in person if she prefers. IAPT measures are routinely collected at each contact so collecting this treatment data will not involve additional burden for women.
All outcomes will be summarized at 3 months postpartum; video and qualitative interview audio data will be collected during a visit to the participant’s home (or remotely) at this point in addition to the IAPT measures.
As per standard practice in IAPT services, anxiety, depression and disorder-specific measures will be taken at each point of contact. Participants will be asked to complete the additional measures at the baseline and outcome points.
Video data
All video recordings will be collected using password-protected iPads or via Microsoft Teams. The iPads will be brought back to the university site for secure storage directly after the interview. Video recordings will be immediately uploaded on to the secure university computer network, which is password-protected and which only the research team have access. Once the video recordings have been transferred onto the secure computer network, they will be permanently deleted from the iPads or TEAMS. In order to verify the deletion of the files, we will empty the ‘trash’ file of the iPads. The iPads will be linked to a cloud account in order to be able to wipe them remotely should they be stolen. The video data files will be saved on the secure university computer network using only participant ID and date as identifiers, and only researchers involved in the project will have access to the drive with the video recordings.
Confidentiality
Participants will be registered as patients under secure local NHS clinical patient records systems (known as IAPTUS) which will hold their personal data. Participants research data (questionnaires) will be pseudonymised and entered on password-protected databases as described above, kept on university computers. Video and audio data will be identified by participants number only. Participants names will be kept in a separate password protected database.
Statistical methods
Quantitative data
Quantitative data analysis will be primarily descriptive to aid the planning of a future RCT. Participant flow through the study will be presented following CONSORT guidelines. Descriptive data will be presented in the form of means and standard deviations; medians and ranges; or percentages with 95% confidence intervals, as appropriate depending on the data being described.
The following feasibility parameters will be calculated: (1) percentage of participants meeting eligibility criteria (those eligible/those approached or referred) by recruitment channel into the study; (2) number of participants recruited per month (defined as those consented); (3) percentage of individuals consenting to randomisation (those randomised/those consented); (4) percentage of participants completing treatment in each arm (number of participants receiving adequate treatment dose, defined as 60% of treatment hours/those randomised); (5) percentage completing the outcome measures at 3m postpartum (number of participants completed at least one follow-up measure/those randomised); (6) between group pre-post effect sizes and confidence intervals, adjusting for randomisation stratifiers (fixed effect) and therapist (random effect), and variance on the potential primary outcome measure at 3 months post-randomisation; (7) summary statistics on acceptability of intervention and outcome measures measured using a 3-point Likert scale. Data will be presented overall and subgroup summaries by disorder.
Descriptive data on therapist competence and compliance will be presented using standardised Cognitive Therapy Scale Ratings (CTS-R) and number of treatment elements completed from checklist.
Qualitative data
Focussed thematic analysis will be utilised, as in a previous study evaluating experiences of treatment [58, 59] which compared participants’ experiences in a non-randomised trial of intensive and weekly CBT for OCD. Constant comparison method [60] will be used to delineate themes and sub-themes relating to participants’ experiences and attitudes towards treatment.
Two researchers will independently code three transcripts to help identify and discuss alternative interpretations of the data [61]. An analytical framework will be constructed around the perceived value, acceptability and feasibility of the treatment, which will be applied to the remaining transcripts, with themes and subthemes refined as necessary. Deductive codes will be supplemented with inductive codes to reflect the emergent priorities and concerns in the data. Ideas about themes and their relationships will be recorded in theoretical memos and discussed amongst our Project Service User Advisory Group. The computer programme QSR N-VIVO will be used to process the transcripts, enabling coding and retrieval of a large volume of narrative data.
Monitoring
Composition of the coordinating centre and trial steering committee
The Trial Management Group (TMG) comprises FC, LP, BC and VL. An independent trial steering and data monitoring committee will be established to examine the clinical progress of trial participants and the conduct of the trial. The group comprises two clinicians, one statistician and one expert by experience. This will meet soon after the beginning of recruitment and then every 6 months to oversee the trial, check data and adverse events; a trial report is prepared in advance. A charter for the group is available from the corresponding author.
Patient and public involvement
Patients were involved in advising on the design of the study. A patient advisory group (PAG) comprising 4–6 women with lived experience of perinatal mental health problems will be set up to advise on the study and will meet approximately annually.
Adverse event reporting and harms
Participants will be carefully monitored throughout treatment by asking them for relevant information at each contact. Data will be collected from participants on potential adverse effects including pregnancy outcomes and prematurity.
Adverse events will be monitored and recorded, including the mental and physical health of the mother and any pregnancy-related events. Should participants deteriorate clinically in terms of significantly increased symptoms of anxiety or depression, or an increase in risk to themselves or others, then the intervention will be stopped and a referral made to specialist perinatal services and/or other services as clinically appropriate.
Dissemination plans
-
1.
Results of the trial will be fed back to participants via a newsletter.
-
2.
Findings from the study will be published in a series of high-quality peer reviewed papers. These will include journals targeted at academics, CBT practitioners, perinatal specialists and health service managers.
-
3.
Study results will be presented at academic and service user led conferences as well as conferences for managers. Examples would be the British Association for Behavioural and Cognitive Psychotherapies conference, Marce conference, Maternal Mental Health Alliance conference.
-
4.
Findings from the study will be disseminated to service user groups in perinatal mental health and for anxiety disorders and umbrella organisations such as the maternal mental health alliance.
-
5.
Clinical approaches developed from the study would be disseminated in clinical skills workshops and training for existing and new CBT therapists. These would take place in IAPT and specialist perinatal settings.
Protocol version and amendments
This publication is based on protocol number and date ADEPT Protocol v3_03.06.20.
Discussion
There is a need for effective and accessible treatments for pregnant women with anxiety disorders. This study will be the first to examine the use of time-intensive CBT in pregnant women. It is designed to assess questions of acceptability and feasibility, in order to inform the design of a full-scale RCT. The study data will inform this using both quantitative and qualitative data to assess feasibility outcomes. Time intensive CBT has the potential to be an effective and time efficient intervention for women with anxiety during pregnancy. It is designed to be delivered in existing psychological services and could therefore potentially be rolled out widely across the NHS.
Trial status
Recruitment started in September 2019 and is expected to be completed by end September 2021.
Availability of data and materials
Not currently applicable. Participant level data will not be available due to the small and potentially identifiable dataset.
Abbreviations
- ADSUS:
-
Adult service use measure
- CADAT:
-
Centre for anxiety disorders and trauma
- CBT:
-
Cognitive behaviour therapy
- CI:
-
Chief investigator
- CTS-R:
-
Cognitive therapy scale-revised
- CTU:
-
Clinical trials unit
- DSM-V:
-
Diagnostic and statistical manual of mental disorders fifth edition
- GAD:
-
Generalised anxiety disorder
- GAD-7:
-
Generalised anxiety disorder scale-7 items
- HI:
-
High intensity
- IES:
-
Impact of events scale
- NHS:
-
National health service
- NICE:
-
National institute for health and care excellence
- IAPT:
-
Improving access to psychological therapies
- OCD:
-
Obsessive-compulsive disorder
- OCI-R:
-
Obsessive compulsive inventory-revised
- PAG:
-
Patient advisory group
- PBQ:
-
Postpartum bonding questionnaire
- PHQ-9:
-
Patient health questionnaire
- PRAQ:
-
Pregnancy-related anxiety questionnaire
- PTSD:
-
Post-traumatic stress disorder
- RCT:
-
Randomised controlled trial
- SCID-V:
-
Structured clinical interview DSM-V
- SLaM:
-
South London and Maudsley NHS Trust
- SPIN:
-
The social phobia inventory
- SPSS:
-
Statistical package for the social sciences
- WSAS:
-
Work and social adjustment scale
- TMG:
-
Trial management group
References
Stein A, Pearson RM, Goodman SH, Rapa E, Rahman A, McCallum M, et al. Effects of perinatal mental disorders on the fetus and child. Lancet. 2014;384(9956):1800–19. https://doi.org/10.1016/S0140-6736(14)61277-0.
Goodman JH, Chenausky KL, Freeman MP. Anxiety disorders during pregnancy: A systematic review. J Clin Psychiatry. 2014;75(10):e1153–e84. https://doi.org/10.4088/JCP.14r09035.
Andersson L, Sundström-Poromaa I, Wulff M, Åström M, Bixo M. Depression and anxiety during pregnancy and six months postpartum: a follow-up study. Acta Obstet Gynecol Scand. 2006;85(8):937–44. https://doi.org/10.1080/00016340600697652.
Fairbrother N, Janssen P, Antony MM, Tucker E, Young AH. Perinatal anxiety disorder prevalence and incidence. J Affect Disord. 2016;200:148–55. https://doi.org/10.1016/j.jad.2015.12.082.
Dennis C-L, Falah-Hassani K, Shiri R. Prevalence of antenatal and postnatal anxiety: systematic review and meta-analysis. Br J Psychiatry. 2017;210(5):315–23. https://doi.org/10.1192/bjp.bp.116.187179.
Martini J, Asselmann E, Einsle F, Strehle J, Wittchen HU. A prospective-longitudinal study on the association of anxiety disorders prior to pregnancy and pregnancy- and child-related fears. J Anxiety Disord. 2016;40:58–66. https://doi.org/10.1016/j.janxdis.2016.04.007.
Ross LE, McLean LM. Anxiety disorders during pregnancy and the postpartum period: a systematic review. J Clin Psychiatry. 2006;67(8):1285–98. https://doi.org/10.4088/JCP.v67n0818.
Martini J, Petzoldt J, Einsle F, Beesdo-Baum K, Hofler M, Wittchen H-U. Risk factors and course patterns of anxiety and depressive disorders during pregnancy and after delivery: a prospective-longitudinal study. J Affect Disord. 2015;175:385–95. https://doi.org/10.1016/j.jad.2015.01.012.
Warren SL, Gunnar MR, Kagan J, Anders TF, Simmens SJ, Rones M, et al. Maternal panic disorder: Infant temperament, neurophysiology, and parenting behaviors. J Am Acad Child Adolesc Psychiatry. 2003;42(7):814–25. https://doi.org/10.1097/01.CHI.0000046872.56865.02.
Murray L, Lau PY, Arteche A, Creswell C, Russ S, Zoppa LD, et al. Parenting by anxious mothers: effects of disorder subtype, context and child characteristics. J Child Psychol Psychiatry. 2012;53(2):188–96. https://doi.org/10.1111/j.1469-7610.2011.02473.x.
Stein A, Craske MG, Lehtonen A, Harvey A, Savage-McGlynn E, Davies B, et al. Maternal cognitions and mother-infant interaction in postnatal depression and generalized anxiety disorder. J Abnorm Psychol. 2012;121(4):795–809. https://doi.org/10.1037/a0026847.
Challacombe FL, Salkovskis PM, Woolgar M, Wilkinson EL, Read J, Acheson R. Parenting and mother-infant interactions in the context of maternal postpartum obsessive-compulsive disorder: Effects of obsessional symptoms and mood. Infant Behav Dev. 2016;44:11–20. https://doi.org/10.1016/j.infbeh.2016.04.003.
O'Connor TG, Heron J, Golding J, Glover V. Maternal antenatal anxiety and behavioural/emotional problems in children: a test of a programming hypothesis. J Child Psychol Psychiatry. 2003;44(7):1025–36. https://doi.org/10.1111/1469-7610.00187.
Glover V. Maternal depression, anxiety and stress during pregnancy and child outcome; what needs to be done. Best Pract Res Clin Obstet Gynaecol. 2014;28(1):25–35. https://doi.org/10.1016/j.bpobgyn.2013.08.017.
Martini J, Knappe S, Beesdo-Baum K, Lieb R, Wittchen H-U. Anxiety disorders before birth and self-perceived distress during pregnancy: associations with maternal depression and obstetric, neonatal and early childhood outcomes. Early Hum Dev. 2010;86(5):305–10. https://doi.org/10.1016/j.earlhumdev.2010.04.004.
Merikangas KR, Avenevoli S, Dierker L, Grillon C. Vulnerability factors among children at risk for anxiety disorders. Biol Psychiatry. 1999;46(11):1523–35. https://doi.org/10.1016/S0006-3223(99)00172-9.
National Institute for Health and Care Excellence (2014). Antenatal and postnatal mental health (NICE Guideline 192). Available at https://www.nice.org.uk/guidance/cg192 [Accessed 31/5/2020]
Tietz A, Zietlow A, Reck C. Maternal bonding in mothers with postpartum anxiety disorder: The crucial role of subclinical depressive symptoms and maternal avoidance behaviour. Arch Womens Ment Health. 2014;17(5):433–42. https://doi.org/10.1007/s00737-014-0423-x.
Arch JJ. Cognitive behavioral therapy and pharmacotherapy for anxiety: treatment preferences and credibility among pregnant and non-pregnant women. Behav Res Ther. 2014;52:53–60. https://doi.org/10.1016/j.brat.2013.11.003.
Department of Health. IAPT perinatal positive practice guide. London. Available at: https://kirkleesiapt.co.uk/wp-content/uploads/2018/05/perinatal-positive-practice-guide-2013.pdf; 2013. p. 18.
National Institute for Health and Care Excellence (2013). Social anxiety disorder: recognition, assessment and treatment (NICE Guideline 159). Available at https://www.nice.org.uk/guidance/cg159 [Accessed 31/5/2020]
National Institute for Health and Care Excellence (2011). Generalised anxiety disorder and panic disorder in adults (NICE Guideline 113). Available at https://www.nice.org.uk/guidance/cg113 [Accessed 31/5/2020]
National Institute for Health and Care Excellence (2005). Obsessive-compulsive disorder and body dysmorphic disorder: treatment (NICE Guideline 31). Available at https://www.nice.org.uk/guidance/cg31 [Accessed 31/5/2020]
Marchesi C, Ossola P, Amerio A, Daniel BD, Tonna M, De Panfilis C. Clinical management of perinatal anxiety disorders: a systematic review. J Affect Disord. 2016;190:543–50. https://doi.org/10.1016/j.jad.2015.11.004.
Jonsson H, Kristensen M, Arendt M. Intensive cognitive behavioural therapy for obsessive-compulsive disorder: A systematic review and meta-analysis. J Obsessive Compulsive Relat Disord. 2015;6:83–96. https://doi.org/10.1016/j.jocrd.2015.04.004.
Ehlers A, Hackmann A, Grey N, Wild J, Liness S, Albert I, et al. A randomized controlled trial of 7-day intensive and standard weekly cognitive therapy for PTSD and emotion-focused supportive therapy. Am J Psychiatry. 2014;171(3):294–304. https://doi.org/10.1176/appi.ajp.2013.13040552.
Mortberg E, Clark DM, Bejerot S. Intensive group cognitive therapy and individual cognitive therapy for social phobia: Sustained improvement at 5-year follow-up. J Anxiety Disord. 2011;25(8):994–1000. https://doi.org/10.1016/j.janxdis.2011.06.007.
Oldfield VB, Salkovskis PM, Taylor T. Time-intensive cognitive behaviour therapy for obsessive-compulsive disorder: A case series and matched comparison group. Br J Clin Psychol. 2011;50(1):7–18. https://doi.org/10.1348/014466510X490073.
Challacombe FL, Salkovskis PM. Intensive cognitive-behavioural treatment for women with postnatal obsessive-compulsive disorder: a consecutive case series. Behav Res Ther. 2011;49(6-7):422–6. https://doi.org/10.1016/j.brat.2011.03.006.
Challacombe FL, Salkovskis PM, Woolgar M, Wilkinson EL, Read J, Acheson R. A pilot randomized controlled trial of time-intensive cognitive-behaviour therapy for postpartum obsessive-compulsive disorder: effects on maternal symptoms, mother-infant interactions and attachment. Psychol Med. 2017;47:1–11.
Chan AW, Tetzlaff JM, Altman DG, Laupacis A, Gotzsche PC, Krleza-Jeric K, et al. SPIRIT 2013 statement: defining standard protocol items for clinical trials. Ann Intern Med. 2013;158(3):200–7. https://doi.org/10.7326/0003-4819-158-3-201302050-00583.
Blackburn IM, James IA, Milne D, Baker C, Standart S, Garland A, et al. The revised cognitive therapy scale (CTS-R): psychometric properties. Behav Cogn Psychother. 2001;29(4):431–46. https://doi.org/10.1017/S1352465801004040.
First MB, Williams JB, Karg RS, Spitzer RL. Structured Clinical Interview for DSM-5—Research Version. Arlington: American Psychiatric Association; 2015.
American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th ed. Arlington: American Psychiatric Publishing; 2013.
Spitzer RL, Kroenke K, Williams JW, Löwe B. A brief measure for assessing generalized anxiety disorder: the GAD-7. Arch Intern Med. 2006;166(10):1092–7. https://doi.org/10.1001/archinte.166.10.1092.
Gyani A, Shafran R, Layard R, Clark DM. Enhancing recovery rates: Lessons from year one of IAPT. Behav Res Ther. 2013;51(9):597–606. https://doi.org/10.1016/j.brat.2013.06.004.
Kroenke K, Spitzer RL, Williams JBW. The PHQ-9: Validity of a brief depression severity measure. J Gen Intern Med. 2001;16(9):606–13. https://doi.org/10.1046/j.1525-1497.2001.016009606.x.
Mundt JC, Marks IM, Shear M, Greist JM. The work and social adjustment scale: a simple measure of impairment in functioning. Br J Psychiatry. 2002;180(5):461–4. https://doi.org/10.1192/bjp.180.5.461.
Foa EB, Huppert JD, Leiberg S, Langner R, Kichic R, Hajcak G, et al. The obsessive-complusive inventory: development and validation of a short version. Psychol Assess. 2002;14(4):485–95. https://doi.org/10.1037/1040-3590.14.4.485.
Chambless DL, Caputo GC, Jasin SE, Gracely EJ, Williams C. The mobility inventory for agoraphobia. Behav Res Ther. 1985;23(1):35–44. https://doi.org/10.1016/0005-7967(85)90140-8.
Weiss DSM, C.R. The Impact of Event Scale-Revised. In: J.P. Wilson TMKE, editor. Assessing Psychological Trauma and PTSD: A Practitioner’s Handbook. New York: Guilford Press; 1997. p. 399–411.
Creamer M, Bell R, Failla S. Psychometric properties of the Impact of Event Scale - Revised. Behav Res Ther. 2003;41(12):1489–96. https://doi.org/10.1016/j.brat.2003.07.010.
Connor KM, Davidson JRT, Churchill LE, Sherwood A, Weisler RH, Foa E. Psychometric properties of the Social Phobia Inventory (SPIN). New self-rating scale. Br J Psychiatry. 2000;176(4):379–86. https://doi.org/10.1192/bjp.176.4.379.
Hatcher RL, Gillaspy JA. Development and validation of a revised short version of the Working Alliance Inventory. Psychother Res. 2006;16(1):12–25. https://doi.org/10.1080/10503300500352500.
Huizink AC, Delforterie MJ, Scheinin NM, Tolvanen M, Karlsson L, Karlsson H. Adaption of pregnancy anxiety questionnaire-revised for all pregnant women regardless of parity: PRAQ-R2. Arch Womens Ment Health. 2016;19(1):125–32. https://doi.org/10.1007/s00737-015-0531-2.
Brockington IF, Fraser C, Wilson D. The postpartum bonding questionnaire: a validation. Arch Womens Ment Health. 2006;9(5):233–42. https://doi.org/10.1007/s00737-006-0132-1.
Crittenden PM. CARE Index manual. Miami: Family Relations Institute; 2003.
Steadman J, Pawlby S, Mayers A, Bucks RS, Gregoire A, Miele-Norton M, et al. An exploratory study of the relationship between mother-infant interaction and maternal cognitive function in mothers with mental illness. J Reprod Infant Psychol. 2007;25(4):255–69. https://doi.org/10.1080/02646830701691343.
Kenny M, Conroy S, Pariante CM, Seneviratne G, Pawlby S. Mother-infant interaction in mother and baby unit patients: Before and after treatment. J Psychiatr Res. 2013;47(9):1192–8. https://doi.org/10.1016/j.jpsychires.2013.05.012.
Conroy S, Pariante CM, Marks MN, Davies HA, Farrelly S, Schacht R, et al. Maternal psychopathology and infant development at 18 months: the impact of maternal personality disorder and depression. J Am Acad Child Adolesc Psychiatry. 2012;51(1):51–61. https://doi.org/10.1016/j.jaac.2011.10.007.
Leventhal A, Jacobsen T, Miller L, Quintana E. Caregiving attitudes and at-risk maternal behavior among mothers with major mental illness. Psychiatr Serv. 2004;55(12):1431–3. https://doi.org/10.1176/appi.ps.55.12.1431.
Trevillion K, Ryan EG, Pickles A, Heslin M, Byford S, Nath S, et al. An exploratory parallel-group randomised controlled trial of antenatal Guided Self-Help (plus usual care) versus usual care alone for pregnant women with depression: DAWN trial. J Affect Disord. 2020;261:187–97. https://doi.org/10.1016/j.jad.2019.10.013.
Saunders B, Sim J, Kingstone T, Baker S, Waterfield J, Bartlam B, Jinks C. Saturation in qualitative research: exploring its conceptualization and operationalization. Qual Quant. 2018;52(4):1893-907.
Roth AD, Pilling S. The competences required to deliver effective cognitive and behavioural therapy for people with depression and with anxiety disorders. London: Department of Health; 2007.
Arain M, Campbell MJ, Cooper CL, Lancaster GA. What is a pilot or feasibility study? A review of current practice and editorial policy. BMC Med Res Methodol. 2010;10(1):67. https://doi.org/10.1186/1471-2288-10-67.
Browne RH. On the use of a pilot sample for sample size determination. Stat Med. 1995;14(17):1933–40. https://doi.org/10.1002/sim.4780141709.
Lancaster GA, Dodd S, Williamson PR. Design and analysis of pilot studies: recommendations for good practice. J Eval Clin Pract. 2004;10(2):307–12. https://doi.org/10.1111/j..2002.384.doc.x.
Bevan A, Oldfield VB, Salkovskis PM. A qualitative study of the acceptability of an intensive format for the delivery of cognitive-behavioural therapy for obsessive-compulsive disorder. Br J Clin Psychol. 2010;49(2):173–91. https://doi.org/10.1348/014466509X447055.
Braun V, Clarke V. Using thematic analysis in psychology. Qualitative Research in Psychology. 2006;3(2):77–101. https://doi.org/10.1191/1478088706qp063oa.
Glaser BG. Theoretical sensitivity. Mill Valley: Sociology Press; 1978.
Tong A, Sainsbury P, Craig J. Consolidated criteria for reporting qualitative research (COREQ): a 32-item checklist for interviews and focus groups. Int J Qual Health Care. 2007;19(6):349–57. https://doi.org/10.1093/intqhc/mzm042.
Acknowledgements
This report is independent research supported by the National Institute for Health Research (HEE/NIHR ICA Programme Clinical Lectureship, Dr Fiona Challacombe, ICA-CL-2017-03-013). This study is supported by the National Institute for Health Research (NIHR) Applied Research Collaboration South London (NIHR ARC South London) at King’s College Hospital NHS Foundation Trust. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care. The NIHR were not involved in the design, data collection, analysis or interpretation of the study.
Funding
This report is independent research supported by the National Institute for Health Research (HEE/NIHR ICA Programme Clinical Lectureship, Dr Fiona Challacombe, ICA-CL-2017-03-013).
Author information
Authors and Affiliations
Contributions
FLC, LP, VL, BC and LMH made substantial contributions to conception and design of the study. FLC, AH and LP drafted the manuscript and all authors provided revisions to the clinical and intellectual content. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
NHS ethics approval was given by London Surrey Borders REC, Reference 19/LO/0622. The trial sponsor is King’s College London. Any protocol amendments will be communicated to all involved parties by email.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Challacombe, F.L., Potts, L., Carter, B. et al. Optimising psychological treatment for Anxiety DisordErs in Pregnancy (ADEPT): study protocol for a feasibility trial of time-intensive CBT versus weekly CBT. Pilot Feasibility Stud 7, 101 (2021). https://doi.org/10.1186/s40814-021-00838-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40814-021-00838-8