
Abstract
Background
Microbes have fundamental roles underpinning the functioning of our planet, they are involved in global carbon and nutrient cycling, and support the existence of multicellular life. The mangrove ecosystem is nutrient limited and if not for microbial cycling of nutrients, life in this harsh environment would likely not exist. The mangroves of Southeast Asia are the oldest and most biodiverse on the planet, and serve vital roles helping to prevent shoreline erosion, act as nursery grounds for many marine species and sequester carbon. Despite these recognised benefits and the importance of microbes in these ecosystems, studies examining the mangrove microbiome in Southeast Asia are scarce.cxs
Results
Here we examine the microbiome of Avicenia alba and Sonneratia alba and identify a core microbiome of 81 taxa. A further eight taxa (Pleurocapsa, Tunicatimonas, Halomonas, Marinomonas, Rubrivirga, Altererythrobacte, Lewinella, and Erythrobacter) were found to be significantly enriched in mangrove tree compartments suggesting key roles in this microbiome. The majority of those identified are involved in nutrient cycling or have roles in the production of compounds that promote host survival.
Conclusion
The identification of a core microbiome furthers our understanding of mangrove microbial biodiversity, particularly in Southeast Asia where studies such as this are rare. The identification of significantly different microbial communities between sampling sites suggests environmental filtering is occurring, with hosts selecting for a microbial consortia most suitable for survival in their immediate environment. As climate change advances, many of these microbial communities are predicted to change, however, without knowing what is currently there, it is impossible to determine the magnitude of any deviations. This work provides an important baseline against which change in microbial community can be measured.
Similar content being viewed by others

Background
Mangrove trees occupy a transitional zone between marine and terrestrial environments, and are the only tree species on the planet that can thrive in the saline, oxygen limited habitat found in this habitat [1,2,3,4]. These highly productive ecosystems are found in tropical and subtropical regions where they have significant ecological and economic importance. They are critical for nutrient cycling, provide basal organic matter to coastal food webs, preventing coastal erosion, filtering pollutants, are biodiversity hotspots, and act as nurseries for many marine animals [5,6,7,8]. Economically, they buffer against natural disasters, support coastal fisheries, and are the source of many forestry products [8, 9].
Microorganisms have key roles in mangrove ecosystems, and are important in promoting growth and maintaining productivity [10,11,12,13]. These microbes contribute significantly to carbon cycling and the global carbon budget [14]. Despite their limited area, accounting for only approximately 3% of global forest cover, mangroves are significant carbon sinks with estimates suggesting they contain 10% of all global carbon emissions [15,16,17]. Known as “blue carbon”, mangroves are able to sequester atmospheric carbon dioxide in above and below ground structures (e.g., leaves and roots etc.). Ultimately, this carbon becomes locked in the anoxic sediments where it remains stable until disturbed [17, 18]. Once disturbed, usually through anthropogenic factors such as habitat clearance or modification, microbial processes can release this carbon back into the atmosphere where it contributes to climate change [19, 20]. However, despite the growing interest in blue carbon, we currently lack a detailed understanding or characterisation of the microbial communities and potential drivers involved in biogeochemical cycling in these coastal ecosystems [21], particularly in Southeast Asia. As climate change progresses and coastal habitats are further degraded by land-use change, eutrophication, and other anthropogenic activities, many of these coastal ecosystems are predicted to become net sources of carbon instead of sinks [19, 20, 22,23,24,25,26]. Consequently, mangroves and other coastal habitats, along with their associated blue carbon stocks, are unlikely to fulfil their claimed potential as nature-based climate solutions, with many of their benefits likely overstated, especially in the light of continued habitat degradation [18, 27,28,29,30,31]
The importance of the microbiome in maintaining and promoting host health has been recognised in many organisms in both terrestrial and marine environments [32,33,34,35,36,37], and the role of the microbiome in promoting plant growth and resilience in terrestrial ecosystems is well established [38, 39]. However, the ecological importance of the coastal microbiome is not yet well-understood, although several studies have shown that fungal and bacterial diversity are key to habitat restoration in marine environments [5, 8]. Several mangrove-associated bacteria are known to promote root growth, support nutrient cycling and availability, degrade contaminants, and aid in other essential processes [5, 7, 40,41,42]. Despite this recognised importance, the dynamics, distribution, and community composition of microbes in mangrove and coastal ecosystems remain vague [10, 11, 43]. This paucity is particularly acute in Southeast Asia (but see [44, 45] for relevant work on seagrasses).
Thirty-four percent of global mangrove cover is found in Southeast Asia, with Indonesia and Malaysia having the largest area of mangrove forest in the region [46,47,48]. Mangroves in Southeast Asia are typically highly productive, and are the oldest and most biodiverse mangrove forests in the world [49,50,51]. Yet, primarily a consequence of anthropogenic activities [52,53,54], regional rates of loss are some of the highest in the world [48, 55, 56]. Restoration of mangrove ecosystems through strategies such as out-planting of nursery-raised saplings, raised bed methods, and direct propagule planting have shown mixed results, with most eventually failing [51, 57,58,59,60]. However, greater success has been achieved when inoculation with local bacterial and fungal species has been performed [5] and the matching of microbial communities between transplant and out-planting sites to mitigate host maladaptation to a new environment has been recommended [61, 62]. Given that microbial communities frequently show distinct compositions, even across comparatively small spatial scales, an understanding of this community structure should be an important consideration in future restoration activities [63,64,65,66,67].
The idea of a ‘core microbiome’ was initially explored by the Human Microbiome Project, and was defined as a group of microbial taxa that are shared by all, or most humans [68]. The ubiquity of these taxa in their host species led to the suggestion that this core microbiome may play an important role in maintaining host biological function [69], and in natural ecosystems, these ubiquitous microbes have similarly been hypothesised to be critical for overall ecological functioning [70, 71]. Here we examine the microbiome of two widespread mangrove species, Sonneratia alba and Avicennia alba throughout the Malay Peninsula. We hypothesise that a ‘core mangrove microbiome’ of shared microbial taxa will exist between both species. More generally, despite sharing a core taxa of microbes throughout the region and between species, we expect to identify microbial community differentiation between species, geographic locations and structure sampled.
Studies such as this are an important step in understanding the Southeast Asian mangrove microbiome, they allow us to generate baseline data on the associated microbial communities. These communities are predicted to change under future projected climate change scenarios [72], but it is impossible to assess the magnitude of these changes, or the taxonomic shifts that will occur without knowing what microbes are present and their spatial structure. As interest in mangrove restoration increases throughout the region, an understanding of the microbes associated with these habitats will become an increasingly important consideration in conservation initiatives, especially as we gain further insights into the vital roles microbes play in promoting and sustaining host health.
Methods
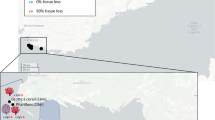
At each of nine sampling locations throughout Singapore and Malaysia, we targeted 10 visibly healthy whole leaves, fruiting bodies (mangrove fruit) and entire pneumatophores from Avicennia alba and Sonneratia alba. In addition to living tissue, a sediment sample was collected in close proximity to each tree (< 1 m), with sediment samples taken from approximately 4 cm below the surface. We were unable to find Avicennia alba at two sample locations in Malaysia (Port Dickson, and Tioman) (Fig. 1). For both species, DNA was extracted with a Qiagen DNeasy PowerSoil Kit (Qiagen, Hilden, Germany) following the manufacturer’s protocol. Prior to extraction all samples were disrupted in an Omni Bead Ruptor 24 (Omni International, Kennesaw, GA, United States) at 8 ms−1 for 2 min. PCR amplification targeting the V4 region of the 16S small sub-unit (SSU) rRNA gene was performed using the 515F and 806R primers modified to include Illumina adaptors, a linker and a unique barcode [73]. All reactions were performed in a total volume of 25 µl, containing 1 µl of undiluted template, 0.1 µl of KAPA 3G Enzyme (Kapa Biosystems, Inc, Wilmington, MA, USA), 0.75 µl of each primer at 10 µM, 2.5 µl, 1.5 µl of 1.5 mg ml−1 BSA, 12.5 µl KAPA PCR Buffer and water to 25 µl. PCR cycling was 94 °C for 180 s, followed by 35 cycles of 94 °C for 45 s, 50 °C for 60 s and 72 °C for 90 s, and a final extension at 72 °C for 10 min. Negative extraction and PCR controls were included to identify any potential contamination issues. Prior to pooling for sequencing, normalisation and cleaning of PCR products was performed in SequalPrep normalisation plates following all manufacturer instructions (Invitrogen). Sequencing was carried out on the Illumina MiSeq platform (600 cycles, V3 chemistry, 300 bp paired end reads) with a 30% PhiX spike (Macrogen).

Map indicating sampling locations
Bioinformatics and statistics
For full details of sequencing statistics and numbers of reads remaining after quality control and filtering, see Additional file 1: Tables S1 and S2. Barcodes and adaptors were removed from de-multiplexed sequence files using Cutadapt version 3.4 [74]. All analysis performed in R used version 3.4.1 (R Core Team, 2017). Reads were filtered based on quality scores and trimmed using the DADA2 package version 1.9.0 [75, 76]. Forward reads were truncated at 300 bp, and reverse reads were truncated at 200 bp. Both forward and reverse reads were filtered to remove any reads less than 100 nucleotides long, or with a max EE (expected error) of 2, and reads were additionally truncated at the end of ‘a good quality sequence’ with the parameter truncQ = 2. The DADA2 algorithm was next used to estimate error rates from all quality-filtered reads and then to merge forward and reverse reads and infer amplicon sequence variants (ASVs). Chimeras were removed with de novo detection. Sequenced extraction and PCR negatives were used to identify possible contaminants using the decontam R package [76], and remaining ASVs were assigned taxonomy with the RDP classifier [77] against a training set based on the Silva v132 16S database [78]. Phylogenetic placement of ASVs was assigned by aligning sequence variants without an anchor using the AlignSeqs() function of the decipher R package version 2.6.0 [79] and constructing a maximum likelihood tree with the optim.pml() function from an initial starting tree built using the NJ() function in the phangorn R package version 2.4.0 [80].
Any ASVs assigned to mitochondrial or chloroplast identities were removed. Raw sequence counts were then converted to relative abundance data. Alpha diversity metrics for each location were calculated, and non-metric multi-dimensional scaling (NMDS) was performed on the UniFrac [81] dissimilarity matrix of samples using vegan version 2.6–4 [82] and phyloseq version 1.41.1 [83] R packages (Fig. 2). Permutational multivariate analysis of variance (PERMANOVA) (Additional file 1: Table S3) was performed on the ASV table with ‘location’ and ‘plant structure’ as predictors using the adonis() function of the vegan package. A Mantel test with 999 permutations was performed using the vegan R package. Differential abundance analyses were performed using the vegan, indicspecies version 1.7.12 [84] and corncob [85] R packages. ASVs and species-level taxa that were detected by all three methods as being differentially abundant in various mangrove structures are reported below. The microbiome R package version 1.10.0 was used to detect a core taxa (Lahti et al. 2017) (Fig. 3). Here we define core taxa as those present in at least 20% of the samples at a relative abundance of at least 10% in each of those samples (i.e., those taxa making up at least 10% of the reads in at least 20% of all samples. These thresholds are inherently arbitrary [86] but were informed by abundance-occupancy distributions [87]. All sequences associated with this work have been deposited at the National Center for Biotechnology Information under BioProject ID: PRJNA735404.

Heatmap showing the abundance of each of the 81 core taxa identified in each species and sampled part

Corncob plot indicating differential abundance of each of the eight taxa identified using three different techniques. Plot only shows the results of corncob analysis. Dots represent model coefficients associated with relative abundance and lines represent 95% prediction intervals for the observed relative abundance (dispersion)
Results
In total, sequencing generated 28,297,986 and 29,484,815 reads for Avicennia alba and Sonneratia alba respectively. After quality control processing and chimera removal, 4,835,163 reads for A. alba and 13,861,503 reads for S. alba remained and were used in all downstream analysis (Additional file 1: Tables S1 and S2). The A. alba and S. alba libraries used in downstream analysis contained a mean of 17,330 (median = 12,785) and 38,944 (median = 38,504) reads per sample respectively, with 12,720 and 24,347 unique ASVs found in each respective library. Rarefaction curves for both species indicate that sufficient sequencing depth was achieved with all samples reaching asymptote (Additional file 1: Fig. S1).
A core microbiome consisting of 81 taxa was identified in both species throughout all samples (Fig. 2). Differential abundance analysis was performed using simper in vegan, indicspecies and corncob. Eight bacterial taxa were identified as differentially abundant between mangrove parts by all three methods. These differentially abundant taxa were Pleurocapsa, Tunicatimonas, Halomonas, Marinomonas, Rubrivirga, Altererythrobacter, Lewinella, and Erythrobacter (Fig. 3). These taxa are key degraders of important marine carbon compounds such as formaldehyde, gelatine, and agar (See discussion), and their significant enrichment in living mangrove parts over sediment (Fig. 3) highlights how important mangroves are as habitats for ecologically critical taxa. PERMANOVA (Additional file 1: Table S3) indicates a weak but significant difference in bacterial communities between host species (p = 0.001, R2 = 0.015), with sampled plant part and sampling location having a larger influence on community structure (p = 0.001, R2 = 0.197; & p = 0.001, R2 = 0.097 respectively). This pattern is also evident in the NMDS plot (Fig. 4) with samples of the same type (e.g., leaves) tending to cluster together irrespective of the host species. Above ground parts (leaves and fruits) tend to be similar while sediment and pneumatophores host bacterial communities that are distinct from each other, the leaves, and the fruits. Further confirming this similarity, plots of beta-dispersion indicate a high degree of overlap in microbial communities between species (Additional file 1: Fig. S2) and between fruits and leaves, while pneumatophores and sediment samples tend to be more dissimilar (Additional file 1: Fig. S3). The highest bacterial diversity was found in sediment samples. In living tissues in both mangrove species, pneumatophores contained the highest diversity, while leaves and fruits showed similar diversity (Additional file 1: Fig. S4). A Mantel test indicated a significant pattern of distance decay of similarity in microbial communities (999 permutations; Mantel statistic r: 0.1097; p = 0.001).

Weighted-UniFrac distance ordination for both species studied and all sampled parts, including sediment
Discussion
Consistent with other work examining microbial community structure in a variety of host taxa, we show that microbial community structure exists around the Malay Peninsula [44, 45, 61, 64, 88, 89], and this structure is primarily a consequence of plant part examined, location sampled, and host identity. It is likely that these differences are driven by local environmental conditions, with locations closer together tending to have more similar environmental conditions in comparison to those that are more geographically distant. This hypothesis is further supported by the significant pattern of distance decay of similarity we observe in microbial communities, meaning that communities are more similar when spatial distance between them is low, a consequence of the host’s ability to exert a degree of control over the composition of their microbial communities, tailoring them to local conditions [90, 91]. Given these findings, and the recognised high rates of failure in mangrove restoration programmes, explicit consideration of microbial communities could help to improve success [92]. More so since inoculation with bacterial and fungal species found in the local environment has already been shown to improve restoration success and promote tolerance to stress [5, 67, 93].
We identified a core Avicennia alba and Sonneratia alba mangrove microbiome consisting of 81 taxa. Given the ubiquity of these taxa in both species and in all living parts (e.g., leaves and pneumatophores) it is likely they play a key role in mediating host fitness. For example, mangroves are nitrogen-limited environments, and in general are considered as nutrient-deficient habitats [94,95,96]. Correspondingly, a number of the taxa we identified in the core microbiome are involved in nitrogen cycling (e.g., Nitrospiraceae nitrospira, Rhizobiales lncertae Sedis Anderseniella, Rhizobiaceae fulvimarina) and thus help promote growth in what would otherwise be a challenging environment for plant life. Similarly, the core microbiome in this work contains a number of taxa involved in sulphur cycling (e.g., Sulfurimonadaceae sulfurimonas & Sulfurovaceae sulfurovum). The bacterial oxidation of sulphur can improve substrate fertility and aid in the removal of toxic sulphide that is produced by sulphur-reducing bacteria [97, 98]. These core taxa would be a good starting point for further investigation as potential inoculates that could improve restoration success, especially when transplants are grown ex-situ. However, increasingly evidence is also suggesting that rare taxa are likely to be just as important [69] and may play important roles in allowing hosts to survive in challenging environments, or in helping facilitate adaption to geographically unique environmental conditions. This is especially true in biogeochemical cycles and in protecting hosts from pathogens [99, 100].
We identified eight bacterial taxa shared between both species that are differentially abundant (Fig. 3). These eight were primarily enriched in living parts suggesting that they play an important role in the Southeast Asian mangrove microbiome. Pleurocapsa, a nitrogen fixing cyanobacteria [101] is enriched within pneumatophores, and it is likely that members of this group facilitate nitrogen uptake in the water-logged and nutrient-limited mangrove sediments.
Members of the Bacteroidetes genus Tunicatimonas (here, enriched in pneumatophores) were first isolated from sea anemones [102] and have since been identified as common on plastic debris in marine environments where they are thought to be well adapted to take advantage of the new niches this plastic creates [103]. While Tunicatimonas might not serve any purpose in promoting host health, their increased abundance could be a consequence of plastic pollution. Plastic pollution is a recognised problem in Southeast Asia, and much of this plastic can become entangled within mangroves where it can disrupt growth and lead to ecological instability [104, 105]. We hypothesize that plastic debris could be facilitating the transport of Tunicatimonas into mangrove ecosystems, particularly since Tunicatimonas taxa are found enriched on pneumatophores, a structure ideally suited to trapping plastic debris.
Halomonas spp. (here, enriched in sediment, fruits, and leaves) have previously been identified in Southeast Asian mangroves where they produce the compound ectoine [106]. This compound is able to stabilize proteins and other cellular structures in the presence of high intensity UV irradiation, heat stresses (cold and high), fluctuations in pH and can help hosts survive extreme osmotic stress [106, 107]. Members of this genus are most enriched in the above ground structures—those that are exposed to the extreme levels of UV light that are frequently encountered in tropical habitats suggesting that they could play a protective role in these parts.
The genus Marinomonas (here, enriched in all living plant tissues) is abundant in marine ecosystems where it is implicated in melanin synthesis and the catabolism of dimethylsulfoniopropionate (DMSP) to dimethyl sulfide (DMS). DMSP metabolisers are acknowledged as important constituents of the coral microbiome where they have principal roles in sulphur cycling, pathogen suppression and mediating thermal stress responses [37, 88, 108,109,110]. The same properties that make DMSP metabolisers beneficial microorganisms in coral mean that this group warrants further investigation in mangroves, particularly as they are enriched in both species. Additionally, this genus is known to degrade agar, a very abundant and recalcitrant carbon compound produced by marine algae.
Little is known of the Rubrivirga genus (here, enriched in leaves and pneumatophores), however, they are described as chemoorganotrophs [111] meaning they are able to oxidise organic chemicals to produce energy. Therefore, they are likely to be involved in nutrient cycling and this genus would be a good candidate for further investigation into its role in the mangrove microbiome.
Similarly, details on Altererythrobacter (here, enriched in leaves and pneumatophores) are scarce, with no information on the specific functions they perform, but they have been isolated from mangrove ecosystems previously [112, 113] and are known to degrade formaldehyde. This ubiquity suggests they have an important role in the mangrove microbiome and would be a good target for further study.
Lewinella (here, enriched in leaves and pneumatophores) are halotolerant heterotrophs that are commonly encountered in activated sludge and are able to break down complex molecules [114] abundant in marine habitats such as gelatine [115]. They show increased abundance when coastal areas undergo sudden vegetation declines that can result from land-use change [116]. Further investigation of this genus may make it possible to identify indicator species that can be used in the design of customised management and conservation strategies.
Erythrobacter (here, enriched in pneumatophores) have been isolated from mangrove habitats in multiple studies throughout the world [95, 117,118,119], and is known to degrade complex molecules such as formaldehyde [120], but little is known about their functions in these habitats. Like Altererythrobacter, the ubiquity of Erythrobacter in multiple mangrove forests suggests that they play an important role in the mangrove microbiome, and again this genus would be a worthy candidate for future studies trying to determine whether beneficial mangrove microbes exist.
Despite having a shared core of 81 taxa across all living parts, each part does host a significantly different bacterial community. In both species, leaves and fruiting bodies, or above ground structures have a similar diversity, and of the living parts, the pneumatophore has the most diverse microbial community. Similar to other studies, the highest diversity of microbes is seen in sediment samples [44, 45, 65, 66]. Our ordinations further support these observations with fruit and leaves having similar microbial communities and therefore clustering together, while pneumatophores and sediment samples form well-defined and clearly differentiated clusters. In all cases, the microbial communities from the same species, in the same sampled part (e.g., A. alba and S. alba leaves) appear similar and do not form distinct clusters. This similarity corroborates the high number of shared core taxa between both species.
Among the eight taxa found to have significantly different abundance, all but one (Halmonas) exhibited strong preference for mangrove plant components over sediment (Fig. 3). This indicates that these potentially important taxa depend on mangroves for their persistence in the coastal environments of Southeast Asia, and further highlights the importance of mangroves for fostering microbial diversity and hosting bacterial taxa that play key roles in nutrient cycling.
Microbes have fundamental roles in the cycling of many elements, and are major regulators of greenhouse gasses [121]. Climate change will alter how these microbial communities are structured [19, 20, 24, 26], and experimental manipulations show that 4 °C of warming can increase soil respiration by as much as 37%, with much of this increase mediated by microbial decomposition of sequestered carbon [24]. Under the anticipated future climate change scenarios, much of the carbon stored in coastal ecosystems will become vulnerable to increased microbial decomposition, and mangrove ecosystems will likely move from net sinks to net sources of greenhouse gasses. Given this, it is crucial that we understand how mangrove microbial communities are currently assembled and structured in order to determine how best to keep existing carbon stores intact.
The arguments in favour of mangrove restoration to increase biodiversity are unequivocal. However, things are much less clear when it comes to blue carbon and how climate change and restoration will impact greenhouse gas fluxes in coastal ecosystems [122,123,124]. Anecdotally, the consensus suggests that restoration will aid in the sequestering of atmospheric carbon. Unfortunately, scientific surveys of greenhouse gas fluxes in restored habitats show that CO2 fluxes in these ecosystems have actually increased. These post restoration increases are observed in numerous coastal ecosystems (e.g., mangroves, kelp forests, saltmarshes and seagrasses) [27, 31, 116, 125,126,127] and demonstrate the challenges involved when trying to understand greenhouse gas fluxes in these ecosystems. These challenges underline the need for studies such as this, without them we will not know what microbes are present or how they will change.
Conclusion
This work uncovers a core mangrove microbiome and identifies a more limited set of eight microbial taxa that are differentially abundant in different compartments in both species of mangroves surveyed. Their elevated abundance suggests possible beneficial properties that could improve restoration success, given this; these would be good candidate taxa for further screening to determine what, if any benefits hosts could derive from their associations with them. Importantly, this work provides a baseline that can be used to measure changes in microbial community structure against, this is valuable because it is predicted that these communities will change in response to climate change. However, it is impossible to assess the magnitude or direction of these changes without knowing what is currently present in the mangrove microbiome.
Natural systems are complex, and if all the factors involved in carbon sequestration and its cycling through mangrove ecosystems are not fully considered, especially how the predicted changes in microbial communities as the climate warms impact CO2 flux, it is possible that we will do more harm than good. Particularly when it comes to using coastal ecosystems in carbon trading schemes and in the offsetting of emissions.
Availability of data and materials
All data associated with this work has been deposited at the National Center for Biotechnology Information under BioProject ID: PRJNA735404. All code for downloading, processing, and reproducing analyses is provided as a versioned GitHub release at: https://zenodo.org/record/7802402#.ZDdnjvZByUk.
References
Lambs L, Muller E, Fromard F. Mangrove trees growing in a very saline condition but not using seawater. Rapid Commun Mass Spectrom. 2008;22:2835–43.
Santini NS, Reef R, Lockington DA, Lovelock CE. The use of fresh and saline water sources by the mangrove Avicennia marina. Hydrobiologia. 2015;745:59–68.
Carugati L, Gatto B, Rastelli E, Lo Martire M, Coral C, Greco S, et al. Impact of mangrove forests degradation on biodiversity and ecosystem functioning. Sci Rep. 2018;8:13298.
Howard RJ, Krauss KW, Cormier N, Day RH, Biagas J, Allain L. Plant-plant interactions in a subtropical mangrove-to-marsh transition zone: effects of environmental drivers. J Veg Sci. 2015;26:1198–211.
Holguin G, Vazquez P, Bashan Y. The role of sediment microorganisms in the productivity, conservation, and rehabilitation of mangrove ecosystems: an overview. Biol Fertil Soils. 2001;33:265–78.
Alongi DM. Bacterial productivity and microbial biomass in tropical mangrove sediments. Microb Ecol. 1988;15:59–79.
Jiang X-T, Peng X, Deng G-H, Sheng H-F, Wang Y, Zhou H-W, et al. Illumina sequencing of 16S rRNA tag revealed spatial variations of bacterial communities in a mangrove wetland. Microb Ecol. 2013;66:96–104.
Allard SM, Costa MT, Bulseco AN, Helfer V, Wilkins LGE, Hassenrück C, et al. Introducing the Mangrove Microbiome Initiative: Identifying Microbial Research Priorities and Approaches To Better Understand, Protect, and Rehabilitate Mangrove Ecosystems. mSystems [Internet]. American Society for Microbiology Journals; 2020 [cited 2020 Oct 21];5. Available from: https://msystems.asm.org/content/5/5/e00658-20
Kathiresan K. Importance of Mangrove Ecosystem. ijms [Internet]. 2012 [cited 2021 Dec 17]; Available from: http://biopublisher.ca/index.php/ijms/article/view/521
Zhuang W, Yu X, Hu R, Luo Z, Liu X, Zheng X, et al. Diversity, function and assembly of mangrove root-associated microbial communities at a continuous fine-scale. npj Biofilms Microbiomes. 2020;6:1–10.
Tavares TCL, Bezerra WM, Normando LRO, Rosado AS, Melo VMM. Brazilian semi-arid mangroves-associated microbiome as pools of richness and complexity in a changing world. Front Microbiol. 2021;12:2485.
Nóbrega MS, Silva BS, Tschoeke DA, Appolinario LR, Calegario G, Venas TM, et al. Mangrove microbiome reveals importance of sulfur metabolism in tropical coastal waters. Sci Total Environ. 2022;813: 151889.
Dhal PK, Kopprio GA, Gärdes A. Insights on aquatic microbiome of the Indian Sundarbans mangrove areas. PLoS ONE. 2020;15:e0221543.
Andreote FD, Jiménez DJ, Chaves D, Dias ACF, Luvizotto DM, Dini-Andreote F, et al. The microbiome of brazilian mangrove sediments as revealed by metagenomics. PLoS ONE. 2012;7:e38600.
Hutchison J, Manica A, Swetnam R, Balmford A, Spalding M. Predicting global patterns in mangrove forest biomass. Conserv Lett. 2014;7:233–40.
Kairo J, Mbatha A, Murithi MM, Mungai F. Total ecosystem carbon stocks of mangroves in Lamu, Kenya; and their potential contributions to the climate change Agenda in the country. Front For Glob Change. 2021;4:151.
Alongi DM. Carbon cycling and storage in mangrove forests. Ann Rev Mar Sci. 2014;6:195–219.
Belshe EF, Mateo MA, Gillis L, Zimmer M, Teichberg M. Muddy waters: unintentional consequences of blue carbon research obscure our understanding of organic carbon dynamics in seagrass ecosystems. Front Mar Sci. 2017;4:125.
Bardgett RD, Freeman C, Ostle NJ. Microbial contributions to climate change through carbon cycle feedbacks. ISME J. 2008;2:805–14.
Zhou Y, Sun B, Xie B, Feng K, Zhang Z, Zhang Z, et al. Warming reshaped the microbial hierarchical interactions. Glob Change Biol. 2021;27:6331–47.
Macreadie PI, Atwood TB, Seymour JR, Fontes MLS, Sanderman J, Nielsen DA, et al. Vulnerability of seagrass blue carbon to microbial attack following exposure to warming and oxygen. Sci Total Environ. 2019;686:264–75.
Bonetti G, Limpert KE, Brodersen KE, Trevathan-Tackett SM, Carnell PE, Macreadie PI. The combined effect of short-term hydrological and N-fertilization manipulation of wetlands on CO2, CH4, and N2O emissions. Environ Pollut. 2022;294: 118637.
Githaiga MN, Frouws AM, Kairo JG, Huxham M. Seagrass removal leads to rapid changes in fauna and loss of carbon. Front Ecol Evol. 2019;7:62.
Hicks Pries CE, Castanha C, Porras RC, Torn MS. The whole-soil carbon flux in response to warming. Science. 2017;355:1420–3.
Romero-Olivares AL, Meléndrez-Carballo G, Lago-Lestón A, Treseder KK. Soil metatranscriptomes under long-term experimental warming and drying: fungi allocate resources to cell metabolic maintenance rather than decay. Front Microbiol. 2019. https://doi.org/10.3389/fmicb.2019.01914.
Rillig MC, Ryo M, Lehmann A, Aguilar-Trigueros CA, Buchert S, Wulf A, et al. The role of multiple global change factors in driving soil functions and microbial biodiversity. Science. 2019;366:886–90.
Liu S, Trevathan-Tackett SM, Jiang Z, Cui L, Wu Y, Zhang X, et al. Nutrient loading decreases blue carbon by mediating fungi activities within seagrass meadows. Environ Res. 2022;212: 113280.
Ricart AM, Ward M, Hill TM, Sanford E, Kroeker KJ, Takeshita Y, et al. Commentary: overstated potential for seagrass meadows to mitigate coastal ocean acidification. Front Mar Sci. 2022. https://doi.org/10.3389/fmars.2022.884857.
Rosentreter JA, Al-Haj AN, Fulweiler RW, Williamson P. Methane and nitrous oxide emissions complicate coastal blue carbon assessments. Glob Biogeochem Cycles. 2021;35:e2020GB006858.
Rosentreter JA, Borges AV, Deemer BR, Holgerson MA, Liu S, Song C, et al. Half of global methane emissions come from highly variable aquatic ecosystem sources. Nat Geosci. 2021;14:225–30.
Van Dam B, Lopes C, Zeller MA, Ribas-Ribas M, Wang H, Thomas H. Overstated potential for seagrass meadows to mitigate coastal ocean acidification. Front Mari Sci. 2021. https://doi.org/10.3389/fmars.2021.729992.
Costantini MS, Medeiros MCI, Crampton LH, Reed FA. Wild gut microbiomes reveal individuals, species, and location as drivers of variation in two critically endangered Hawaiian honeycreepers. PeerJ. 2021;9:e12291.
Marchioro GM, Glasl B, Engelen AH, Serrão EA, Bourne DG, Webster NS, et al. Microbiome dynamics in the tissue and mucus of acroporid corals differ in relation to host and environmental parameters. PeerJ. 2020;8:e9644.
Alsaffar Z, Pearman JK, Cúrdia J, Ellis J, Calleja ML, Ruiz-Compean P, et al. The role of seagrass vegetation and local environmental conditions in shaping benthic bacterial and macroinvertebrate communities in a tropical coastal lagoon. Sci Rep. 2020;10:13550.
Epstein HE, Smith HA, Torda G, van Oppen MJ. Microbiome engineering: enhancing climate resilience in corals. Front Ecol Environ. 2019;17:100–8.
Wainwright BJ, Zahn GL, Afiq-Rosli L, Tanzil JTI, Huang D. Host age is not a consistent predictor of microbial diversity in the coral Porites lutea. Sci Rep. 2020;10:14376.
Quek ZBR, Tanzil JTI, Jain SS, Yong WLO, Yu DCY, Soh Z, et al. Limited influence of seasonality on coral microbiomes and endosymbionts in an equatorial reef. Ecol Ind. 2023;146: 109878.
Trivedi P, Leach JE, Tringe SG, Sa T, Singh BK. Plant–microbiome interactions: from community assembly to plant health. Nat Rev Microbiol. 2020;18:1–15.
Moora M, Öpik M, Sen R, Zobel M. Native arbuscular mycorrhizal fungal communities differentially influence the seedling performance of rare and common Pulsatilla species. Funct Ecol. 2004;18:554–62.
Cheung MK, Wong CK, Chu KH, Kwan HS. Community Structure, dynamics and interactions of bacteria, archaea and fungi in subtropical coastal wetland sediments. Sci Rep. 2018;8:1–14.
Santos HF, Carmo FL, Paes JES, Rosado AS, Peixoto RS. Bioremediation of mangroves impacted by petroleum. Water Air Soil Pollut. 2011;216:329–50.
Reis CRG, Nardoto GB, Oliveira RS. Global overview on nitrogen dynamics in mangroves and consequences of increasing nitrogen availability for these systems. Plant Soil. 2017;410:1–19.
Purahong W, Sadubsarn D, Tanunchai B, Wahdan SFM, Sansupa C, Noll M, et al. First Insights into the Microbiome of a Mangrove Tree Reveal Significant Differences in Taxonomic and Functional Composition among Plant and Soil Compartments. Microorganisms [Internet]. 2019 [cited 2020 Jun 9];7. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6955992/
Rabbani G, Yan BC, Lee NLY, Ooi JLS, Lee JN, Huang D, et al. Spatial and structural factors shape seagrass-associated bacterial communities in Singapore and peninsular Malaysia. Front Mar Sci. 2021;8: 659180.
Yan B, Rabbani G, Lee N, Ooi J, Lee J, Huang D, et al. The microbiome of the seagrass Halophila ovalis: community structuring from plant parts to regional scales. Aquat Microb Ecol [Internet]. 2021 [cited 2021 Oct 7]; Available from: https://www.int-res.com/prepress/a01976.html
Romañach SS, DeAngelis DL, Koh HL, Li Y, Teh SY, Raja Barizan RS, et al. Conservation and restoration of mangroves: Global status, perspectives, and prognosis. Ocean Coast Manag. 2018;154:72–82.
Gandhi S, Jones TG. Identifying mangrove deforestation hotspots in South Asia. Southeast Asia and Asia-Pacific Remote Sensing. 2019;11:728.
Thomas N, Lucas R, Bunting P, Hardy A, Rosenqvist A, Simard M. Distribution and drivers of global mangrove forest change, 1996–2010. PLoS ONE. 2017;12:e0179302.
Wee AKS, Mori GM, Lira CF, Núñez-Farfán J, Takayama K, Faulks L, et al. The integration and application of genomic information in mangrove conservation. Conserv Biol. 2019;33:206–9.
Ellison AM, Farnsworth EJ, Merkt RE. Origins of mangrove ecosystems and the mangrove biodiversity anomaly. Glob Ecol Biogeogr. 1999;8:95–115.
Ellison AM. Mangrove restoration: Do we know enough? Restor Ecol. 2000;8:219–29.
Bunting P, Rosenqvist A, Hilarides L, Lucas RM, Thomas N, Tadono T, et al. Global Mangrove Extent Change 1996–2020: Global Mangrove Watch Version 3.0. 2022;32.
Farnsworth EJ, Ellison AM. The global conservation status of mangroves. Ambio. 1997;26:328–34.
Richards DR, Friess DA. Rates and drivers of mangrove deforestation in Southeast Asia, 2000–2012. PNAS. 2016;113:344–9.
Friess DA, Rogers K, Lovelock CE, Krauss KW, Hamilton SE, Lee SY, et al. The state of the world’s mangrove forests: past, present, and future. Annu Rev Environ Resour. 2019;44:89–115.
Murray NJ, Worthington TA, Bunting P, Duce S, Hagger V, Lovelock CE, et al. High-resolution mapping of losses and gains of Earth’s tidal wetlands. Science. 2022;376:744–9.
Bayraktarov E, Saunders MI, Abdullah S, Mills M, Beher J, Possingham HP, et al. The cost and feasibility of marine coastal restoration. Ecol Appl. 2016;26:1055–74.
Duncan C, Primavera JH, Pettorelli N, Thompson JR, Loma RJA, Koldewey HJ. Rehabilitating mangrove ecosystem services: A case study on the relative benefits of abandoned pond reversion from Panay Island. Philippines Mar Pollut Bull. 2016;109:772–82.
Gedan KB, Silliman BR. Using facilitation theory to enhance mangrove restoration. AMBIO J Hum Environ. 2009;38:109–109.
Samson MS, Rollon RN. Growth performance of planted mangroves in the Philippines: revisiting forest management strategies. Ambio. 2008;37:234–40.
Quek ZBR, Zahn G, Lee NLY, Ooi JLS, Lee JN, Huang D, et al. Biogeographic structure of fungal communities in seagrass Halophilia ovalis across the Malay Peninsula. Environ Microbiol Rep. 2021;1758–222913003.
Wainwright BJ, Zahn GL, Zushi J, Lee NLY, Ooi JLS, Lee JN, et al. Seagrass-associated fungal communities show distance decay of similarity that has implications for seagrass management and restoration. Ecol Evol. 2019;9:11288–97.
Wainwright BJ. Spatial and structural factors shape seagrass-associated bacterial communities in Singapore and peninsular Malaysia. Front Mar Sci. 2021;8:11.
Wainwright BJ, Afiq-Rosli L, Zahn GL, Huang D. Characterisation of coral-associated bacterial communities in an urbanised marine environment shows strong divergence over small geographic scales. Coral Reefs. 2019. https://doi.org/10.1007/s00338-019-01837-1.
Lee NLY, Huang D, Quek ZBR, Lee JN, Wainwright BJ. Mangrove-associated fungal communities are differentiated by geographic location and host structure. Front Microbiol. 2019. https://doi.org/10.3389/fmicb.2019.02456/full.
Lee NLY, Huang D, Quek ZBR, Lee JN, Wainwright BJ. Distinct fungal communities associated with different organs of the mangrove Sonneratia alba in the Malay Peninsula. IMA Fungus. 2020;11:17.
Zahn G, Amend AS. Foliar microbiome transplants confer disease resistance in a critically-endangered plant. PeerJ. 2017;5: e4020.
Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature. 2007;449:804–10.
Risely A. Applying the core microbiome to understand host–microbe systems. J Anim Ecol. 2020;89:1549–58.
Bourne DG, Morrow KM, Webster NS. Insights into the coral microbiome: underpinning the health and resilience of reef ecosystems. Annu Rev Microbiol. 2016;70:317–40.
Trevathan-Tackett SM, Sherman CDH, Huggett MJ, Campbell AH, Laverock B, Hurtado-McCormick V, et al. A horizon scan of priorities for coastal marine microbiome research. Nat Ecol Evol. 2019;3:1509–20.
Cavicchioli R, Ripple WJ, Timmis KN, Azam F, Bakken LR, Baylis M, et al. Scientists’ warning to humanity: microorganisms and climate change. Nat Rev Microbiol. 2019;17:569–86.
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. PNAS. 2011;108:4516–22.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011;17:10–2.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3.
Davis NM, Proctor DM, Holmes SP, Relman DA, Callahan BJ. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome. 2018;6:226.
Cole JR, Chai B, Farris RJ, Wang Q, Kulam-Syed-Mohideen AS, McGarrell DM, et al. The ribosomal database project (RDP-II): introducing myRDP space and quality controlled public data. Nucleic Acids Res. 2007;35:D169–72.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–6.
Wright ES. Using DECIPHER v2.0 to analyze big biological sequence data in R. R J. 2016;8:352–9.
Schliep KP. phangorn: phylogenetic analysis in R. Bioinformatics. 2011;27:592–3.
Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R. UniFrac: an effective distance metric for microbial community comparison. ISME J. 2011;5:169–72.
Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, et al. vegan: Community Ecology Package [Internet]. 2019 [cited 2019 Aug 21]. Available from: https://CRAN.R-project.org/package=vegan
McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE. 2013;8: e61217.
Cáceres MD, Legendre P. Associations between species and groups of sites: indices and statistical inference. Ecology. 2009;90:3566–74.
Martin BD, Witten D, Willis AD. Modeling microbial abundances and dysbiosis with beta-binomial regression. Ann Appl Stat. 2020;14:94–115.
Neu AT, Allen EE, Roy K. Defining and quantifying the core microbiome: challenges and prospects. Proc Natl Acad Sci. 2021;118:e2104429118.
Shade A, Stopnisek N. Abundance-occupancy distributions to prioritize plant core microbiome membership. Curr Opin Microbiol. 2019;49:50–8.
Kanisan DP, Quek ZBR, Oh RM, Afiq-Rosli L, Lee JN, Huang D, et al. Diversity and distribution of microbial communities associated with reef corals of the Malay Peninsula. Microb Ecol. 2022. https://doi.org/10.1007/s00248-022-01958-1.
Oh RM, Bollati E, Maithani P, Huang D, Wainwright BJ. The Microbiome of the reef macroalga sargassum ilicifolium in Singapore. Microorganisms. 2021;9:898.
Goldmann K, Schröter K, Pena R, Schöning I, Schrumpf M, Buscot F, et al. Divergent habitat filtering of root and soil fungal communities in temperate beech forests. Sci Rep. 2016;6:1–10.
Jones P, Garcia BJ, Furches A, Tuskan GA, Jacobson D. Plant host-associated mechanisms for microbial selection. Front Plant Sci. 2019. https://doi.org/10.3389/fpls.2019.00862/full.
Gomes NC, Cleary DF, Calado R, Costa R. Mangrove bacterial richness. Commun Integr Biol. 2011;4:419–23.
Li J, Wang J, Liu H, Macdonald CA, Singh BK. Application of microbial inoculants significantly enhances crop productivity: A meta-analysis of studies from 2010 to 2020. J Sustain Agric Environ. 2022. https://doi.org/10.1002/sae2.12028.
Fernandes SO, Bonin PC, Michotey VD, Garcia N, LokaBharathi PA. Nitrogen-limited mangrove ecosystems conserve N through dissimilatory nitrate reduction to ammonium. Sci Rep. 2012;2:419.
Zilius M, Bonaglia S, Broman E, Chiozzini VG, Samuiloviene A, Nascimento FJA, et al. N2 fixation dominates nitrogen cycling in a mangrove fiddler crab holobiont. Sci Rep. 2020;10:13966.
Zhang Y, Yang Q, Ling J, Van Nostrand JD, Shi Z, Zhou J, et al. Diversity and structure of diazotrophic communities in mangrove rhizosphere, revealed by high-throughput sequencing. Front Microbiol. 2017. https://doi.org/10.3389/fmicb.2017.02032.
Behera BC, Mishra R, Dutta SK, Thatoi HN. Sulphur oxidising bacteria in mangrove ecosystem: a review. Afr J Biotechnol. 2014;13:2897–907.
Varon-Lopez M, Dias ACF, Fasanella CC, Durrer A, Melo IS, Kuramae EE, et al. Sulphur-oxidizing and sulphate-reducing communities in Brazilian mangrove sediments. Environ Microbiol. 2014;16:845–55.
Jousset A, Bienhold C, Chatzinotas A, Gallien L, Gobet A, Kurm V, et al. Where less may be more: how the rare biosphere pulls ecosystems strings. ISME J. 2017;11:853–62.
Hol WHG, de Boer W, de Hollander M, Kuramae EE, Meisner A, van der Putten WH. Context dependency and saturating effects of loss of rare soil microbes on plant productivity. Front Plant Sci. 2015. https://doi.org/10.3389/fpls.2015.00485.
Waterbury JB, Stanier RY. Patterns of growth and development in pleurocapsalean cyanobacteria. Microbiol Rev. 1978;42:2–44.
Yoon J, Oku N, Park S, Katsuta A, Kasai H. Tunicatimonas pelagia gen. nov., sp. nov., a novel representative of the family Flammeovirgaceae isolated from a sea anemone by the differential growth screening method. Antonie Van Leeuwenhoek. 2012;101:133–40.
Bryant JA, Clemente TM, Viviani DA, Fong AA, Thomas KA, Kemp P, et al. Diversity and Activity of communities inhabiting plastic debris in the north pacific gyre. mSystems. 2016;1:e00024-e116.
Rahim S, Widayati W, Analuddin K, Saleh F, Sahar S. Spatial distribution of marine debris pollution in mangrove-estuaries ecosystem of Kendari Bay. IOP Conf Ser Earth Environ Sci. 2020;412:012006.
Adyel TM, Macreadie PI. World’s largest mangrove forest becoming plastic cesspit. Front Mar Sci. 2021. https://doi.org/10.3389/fmars.2021.766876.
Van Thuoc D, Hien TT, Sudesh K. Identification and characterization of ectoine-producing bacteria isolated from Can Gio mangrove soil in Vietnam. Ann Microbiol BioMed Central. 2019;69:819–28.
Lippert K, Galinski EA. Enzyme stabilization be ectoine-type compatible solutes: protection against heating, freezing and drying. Appl Microbiol Biotechnol. 1992;37:61–5.
Raina J-B, Tapiolas DM, Forêt S, Lutz A, Abrego D, Ceh J, et al. DMSP biosynthesis by an animal and its role in coral thermal stress response. Nature. 2013;502:677–80.
Peixoto RS, Rosado PM, de Leite DCA, Rosado AS, Bourne DG. Beneficial microorganisms for corals (BMC): proposed mechanisms for coral health and resilience. Front Microbiol. 2017. https://doi.org/10.3389/fmicb.2017.00341/full.
Lucas-Elío P, Goodwin L, Woyke T, Pitluck S, Nolan M, Kyrpides NC, et al. Complete genome sequence of Marinomonas posidonica type strain (IVIA-Po-181T). Stand Genomic Sci BioMed Central. 2012;7:31–43.
Goh KM, Chan K-G, Lim SW, Liew KJ, Chan CS, Shamsir MS, et al. Genome analysis of a new rhodothermaceae strain isolated from a hot spring. Front Microbiol. 2016. https://doi.org/10.3389/fmicb.2016.01109.
Liao H, Li Y, Zhang M, Lin X, Lai Q, Tian Y. Altererythrobacter mangrovi sp. nov., isolated from mangrove sediment. Int J Syst Evol Microbiol. 2017;67:4851–6.
Ma H, Ren H, Huang L, Luo Y. Altererythrobacter flavus sp. nov., isolated from mangrove sediment. Int J Syst Evol Microbiol. 2018;68:2265–70.
Kang H, Kim H, Joung Y, Joh K. Lewinella maritima sp. nov., and Lewinella lacunae sp. nov., novel bacteria from marine environments. Int J System Evol Microbiol. 2017;67:3603–9.
Barberán A, Caceres Velazquez H, Jones S, Fierer N. Hiding in plain sight: mining bacterial species records for phenotypic trait information. mSphere. 2017;2:e00237-e317.
Elmer WH, Thiel P, Steven B. Response of sediment bacterial communities to sudden vegetation dieback in a Coastal Wetland. Phytobiomes J. 2017;1:5–13.
Ye Y-H, Anwar N, Xamxidin M, Zhang R, Yan C, Nie Y-F, et al. Description of Erythrobacter mangrovi sp. nov., an aerobic bacterium from rhizosphere soil of mangrove plant (Kandelia candel). Antonie Van Leeuwenhoek. 2020;113:1425–35.
Lei X, Zhang H, Chen Y, Li Y, Chen Z, Lai Q, et al. Erythrobacter luteus sp. nov., isolated from mangrove sediment. Int J Syst Evol Microbiol. 2015;65:2472–8.
Alzubaidy H, Essack M, Malas TB, Bokhari A, Motwalli O, Kamanu FK, et al. Rhizosphere microbiome metagenomics of gray mangroves (Avicennia marina) in the Red Sea. Gene. 2016;576:626–36.
Chang A, Jeske L, Ulbrich S, Hofmann J, Koblitz J, Schomburg I, et al. BRENDA, the ELIXIR core data resource in 2021: new developments and updates. Nucleic Acids Res. 2021;49:D498-508.
Tiedje JM, Bruns MA, Casadevall A, Criddle CS, Eloe-Fadrosh E, Karl DM, et al. Microbes and climate change: a research prospectus for the future. mBio. 2022;0:e00800–22.
Macreadie PI, Costa MDP, Atwood TB, Friess DA, Kelleway JJ, Kennedy H, et al. Blue carbon as a natural climate solution. Nat Rev Earth Environ. 2021;1–14.
Macreadie PI, Anton A, Raven JA, Beaumont N, Connolly RM, Friess DA, et al. The future of Blue Carbon science. Nat Commun. 2019;10:3998.
Williamson P, Gattuso J-P. Carbon removal using coastal blue carbon ecosystems is uncertain and unreliable, with questionable climatic cost-effectiveness. Front Clim. 2022. https://doi.org/10.3389/fclim.2022.853666.
Call M, Santos IR, Dittmar T, de Rezende CE, Asp NE, Maher DT. High pore-water derived CO2 and CH4 emissions from a macro-tidal mangrove creek in the Amazon region. Geochim Cosmochim Acta. 2019;247:106–20.
Gallagher JB, Shelamoff V, Layton C. Seaweed ecosystems may not mitigate CO2 emissions. ICES J Mar Sci. 2022;fsac011.
Bahlmann E, Weinberg I, Lavrič JV, Eckhardt T, Michaelis W, Santos R, et al. Tidal controls on trace gas dynamics in a seagrass meadow of the Ria Formosa lagoon (southern Portugal). Biogeosciences Copernicus GmbH. 2015;12:1683–96.
Acknowledgements
Collections from Singapore were made under permit number NP/RP18-035 issued by the National Parks Board of Singapore. Collections from Malaysia were made under permit JTLM 630-7Jld.9(9).
Funding
This study was funded Yale-NUS Grants IG20-SI003 & A-0007210-00-00. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Study conception and design, BJW. Sample collection, BJW, JNL. Laboratory work BJW. Analysis, BJW, TM, LB, KH, LS, ZYY, GZ. First draft, BJW, GZ. All authors commented and contributed to improving the manuscript and preparing the manuscript for submission. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not required.
Consent for publication
Not required.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wainwright, B.J., Millar, T., Bowen, L. et al. The core mangrove microbiome reveals shared taxa potentially involved in nutrient cycling and promoting host survival. Environmental Microbiome 18, 47 (2023). https://doi.org/10.1186/s40793-023-00499-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40793-023-00499-5