
Abstract
Pollinators, including solitary bees, are drastically declining worldwide. Among the factors contributing to this decline, bee pathogens and different land uses are of relevance. The link between the gut microbiome composition and host health has been recently studied for social pollinators (e.g. honeybees), whereas the information related to solitary bees is sparse. This work aimed at the characterization of the gut microbiome of the solitary bees Xylocopa augusti, Eucera fervens and Lasioglossum and attempted to correlate the gut microbial composition with the presence and load of different pathogens and land uses. Solitary bees were sampled in different sites (i.e. a farm, a natural reserve, and an urban plant nursery) showing different land uses. DNA was extracted from the gut, 16S rRNA gene amplified and sequenced. Eight pathogens, known for spillover from managed bees to wild ones, were quantified with qPCR. The results showed that the core microbiome profile of the three solitary bees significantly varied in the different species. Pseudomonas was found as the major core taxa in all solitary bees analyzed, whereas Lactobacillus, Spiroplasma and Sodalis were the second most abundant taxa in X. augusti, E. fervens and Lasioglossum, respectively. The main pathogens detected with qPCR were Nosema ceranae, Nosema bombi and Crithidia bombi, although differently abundant in the different bee species and sampling sites. Most microbial taxa did not show any correlation with the land use, apart from Snodgrassella and Nocardioides, showing higher abundances on less anthropized sites. Conversely, the pathogens species and load strongly affected the gut microbial composition, with Bifidobacterium, Apibacter, Serratia, Snodgrassella and Sodalis abundance that positively or negatively correlated with the detected pathogens load. Therefore, pathogens presence and load appear to be the main factor shaping the gut microbiome of solitary bees in Argentina.
Similar content being viewed by others

Background
Pollinators represent a diverse and heterogeneous group of insects with an important role in the ecosystem, since they are responsible for the pollination of 75% of globally important crops [1]. Among pollinators, bees represent a monophyletic group, which include, besides the common honey bee (Apis mellifera), more than 20.000 species belonging to different genera [2]. Differently from most insects, bee biodiversity peaks in temperate areas but very few are present in the tropical ones [3]. Often, non-Apis bees are more effective pollinators than honey bees for both wild flora and crops [4, 5]. The contribution of wild pollinators should not be underestimated because they can fully pollinate crops without the presence of any kind of managed pollinators [6, 7] although it was also demonstrated that the highest efficiency in crop pollination is achieved when wild pollinators and managed bees are co-habiting the same ecological niche [8]. The Buenos Aires province, in Argentina, is one of the most important areas for farming [9] and Ramello et al. [10] found up to 90 different species of wild bees in that province. Among them, Xylocopa augusti, Eucera fervens and bees of the Halictidae family are known for their role as wild pollinators. The Xylocopa genus (Xylocopinae: Xylocopini) includes more than 470 species all over the world [2], and X. augusti is one of the most prevalent in South America [11, 12]. In contrast to other bees, X. augusti can use buzz-pollination visiting a wide range of plant species to obtain pollen and nectar [13,14,15]. Bees from the family (Hymenoptera: Halictidae) are small with a cosmopolitan distribution and over 4.000 species have been described worldwide [15]. Particularly, Lasioglossum and Haictillus genera group some important native wildflowers visitors in Argentina [16] with a solitary behavior. Ramello et al., [10] found up to 21 different species of halictids, nevertheless the identification at genus and species level is arduous with morphometric tools posing a taxonomical issue in the research activities [17,18,19]. Finally, Buenos Aires province also harbors wild bees belonging to the Eucerini tribe (with more than 780 species distributed worldwide) [2] with importance for the agricultural systems, such as the species Eucera fervens [20] (formerly known as Peponapis fervens, subgenera Peponapis [21]). Bees belonging to the Peponapis subgenera are native from South America [22, 23], and usually their pollination activity rely mainly upon plants of the genus Cucurbita [24] and especially on pumpkins [10].
It is known that pollinators are drastically declining worldwide [25, 26]. Different factors are contributing to this decline, like habitat loss, climate change, urbanization, agricultural intensification, pathogens spread at global level [27,28,29], all complex variables that may act in synergy [30]. Bee pathogens show lethal and sublethal effects on bees generally reducing their lifespan [31] and among pathogens, parasites have been identified as main drivers of wild bees decline [32, 33]. Recent works have focused on the relationship between landscape and bee pathogens, and especially vegetation cover (with different degrees of biodiversity) was reported as a determining factor in the parasites and pathogens spread and dynamics [34].
Commensal microbial communities present in bees gut were recently positively correlated with the host growth and health [35, 36]. In healthy bees, microbial communities are carrying out fundamental functions, such as the support in nutrient acquisition, the regulation of immune responses or defense against pathogens and parasites [37, 38], and detoxification of xenobiotics mainly deriving from agriculture [39]. Changes in the gut microbiome composition may have negative consequences in the host, affecting bees’ health and fitness [40]. Most of the bee microbiome studies regard A. mellifera species and little information are available on wild pollinators. Moreover, little is known about the relationship between landscape, pathogens, and bee gut microbial communities. Recently published research reported that both internal and external pathogens are drives the gut microbial community [38, 41], but also environment can deeply shape the gut microbiome [42].
In this study three solitary wild bee taxa, X. augusti, E. fervens and Lasioglossum, with an important role in crop pollination in the Buenos Aires Province, were selected to investigate the gut microbiome composition and the influence of land use and pathogens on the gut microbiome. The questions on which this study is focused are: (i) does the gut microbiome vary in the different bee taxa?; (ii) are there variations in the distribution and amount of the different bee pathogens in the studied bee species?; (iii) what is the effect of land use on the microbiome composition and biotic stresses distribution? (iv) are the core gut microorganisms and the pathogens load related?
Methodology
Study sites
For this study, bee capture sampling was carried out in 3 different sites characterized by different land use and landscape. The first site was located in Santa Paula’s Farm (SP) and it is specialized on Kiwi fruit and Cucurbitaceae vegetables production (route 226, km 10, Mar del Plata, Buenos Aires; 37° 56´ 0.69´´ S; 57° 40´ 40.53´´ W), the second selected site was a Natural Reserve of Buenos Aires Province, Reserva Natural Paititi (RP), with native grasslands and organic agriculture (37° 54′ 47.774'' S, 57° 48′ 44.806'' W), and, finally, a sampling site was a plant nursery, Vivero Antoniucci (VA), with flowering garden bushes and trees (38° 1′ 42.014''S, 57° 37′ 59.374'' W). In all the sapling sites a wide variety of bee species were present including the domestic bee Apis mellifera.
Spatial analysis
For the spatial characterization of the sampling zones, satellite images from the Landsat 8 satellite were used. These images were taken from the Google Earth Engine repository, with atmospheric corrections made under the name "'LANDSAT/LC08/C01/T1_TOA'". Using the Google Earth Engine platform, the free-access satellite images corresponding to the dates on which the sampling was carried out were downloaded. Only those images with a cloud cover of less than 20% were considered. From the images obtained, a representative image of the median of the filtered images was constructed, thus making it possible to work with a single image containing information from the entire sampling period. This image was exported and processed on the freely available software QGIS (https://qgis.org/) where the normalized difference between the infrared and red bands was calculated, an operation that gives a result known as NDVI, which indicates the predominance or not of vegetation by assigning pixel values between 0 and 1. The NDVI index is considered as a good indicator of the physical properties of the vegetation cover such as, leaf area index, vegetation condition and biomass [43]. To complete the spatial characterization, information on land use available in the repository of the Geographic Information System of the province of Buenos Aires "urbasig" (https://www.urbasig.gob.gba.gob.ar) were used. Three points corresponding to the three sampling zones were added to the final images in QGIS, from which a 3 km radius buffer was constructed according to the foraging range of the different species. Figure 1 summarizes the images detailed above.

Spatial Characterization. The figure on the top represents the three sampling sites (A = Reserva Natural Reserva Natural Paititi; B = Santa Paula; C = Vivero Antoniucci). The three figures in the middle with different shades of red shows the different land uses used in the area with the respective references on the left according to the Argentinian national database on land use. Finally, the three figures on the bottom shows the map constructed from the NDVI values, the shades of green correspond to: dark green correspond to a high absorbance of UV light (healthy vegetation cover); light green correspond to lower absorbance (tilled soil, buildings, and roads)
Experimental design
The work includes the spatial analysis of three different sites, the sampling of solitary pollinators in the study sites, the characterization of the pollinator microbiome and the detection, in each sampled individual of 10 different pathogens known to be dispersed by social bees to solitary pollinators. These data are collected in order to correlate the growing anthropization with a greater presence of pathogens and a consequent imbalance of the microbiota.
Bee sampling and DNA extraction
Different collection campaigns were carried out in each site in the period between December and April of the years 2019 and 2020 (corresponding to the Spring–Summer-Autumn season). For each collection sites, three transects of 70 m were delimited, then the collection was performed in a time interval of 30 min per transect, during sunny days between 9 a.m and 1 p.m. Bees were captured directly from flowers with the use of a homemade bee vacuum.
In total 56 bees were collected, kept separately in plastic vials and maintained at -20 ºC until further analysis. Bees identification through morphometric analysis on the head, wings, body structure and genitals was carried out under a stereo microscope with a × 40 magnification. Total genomic DNA was individually extracted from sampled guts using the High Pure PCR Template Preparation kit (Roche Diagnostics) according to [44]. Extracted DNA was used for bees identification through COI (Cytochrome Oxidase subunit I) gene amplification and sequencing, microbiome study and pathogens analyses. COI amplification and Sanger sequencing of amplified amplicons has been performed following the same protocol of Fernandez de Landa et al. [20].
Pathogen identification and quantification
The following pathogens, considered to be the most common for spill-over between different bee species co-populating the study area, were analyzed through qPCR (StepOne™ Real-Time PCR System, Applied Biosystems) according to [44] relaying on gut extracted DNA: N. ceranae, N. apis, N. bombi, Crithidia bombi, Lotmaria passim, Apicystis bombi, Ascosphaera spp. and Apis mellifera Filamentous Virus (AmFV). All qPCR reactions were performed with qPCRBIO SyGreen Mix Hi-ROX (Ca. number PB20.12–20, PCRBIO systems, London, UK), following the manufacturer’s instructions. DNA samples were diluted 1:10 prior to loading in the reaction well. The specific primers used, the melting temperature and the reaction efficiency are reported in Additional file 1: Table S1. Standard curves were constructed using PCR products of the target amplicon gene. The PCR products were purified, quantified with Qubit™ dsDNA HS Assay Kit (Thermo Fisher, Milan, Italy) and the number of copies was determined based on the amplicon length and total concentration (ng/µl). Amplicons were serially diluted to obtain standards ranging from 104 to 108 gene copies. qPCR data output was standardized for reaction efficiency and melting temperature was verified for every sample and target. To better compare the intensity of each pathogen infecting solitary bees, only bees with a positive Ct value showed by qPCR analysis were considered for further analysis. Finally, according to [44], obtained raw data for the analyzed Nosema species, were corrected by copy number. For N. ceranae and N. bombi data are expressed as N. ceranae Units (NcU) and N. bombi Units (NbU).
Gut microbiome and bioinformatic analysis
The microbiome analysis on sampled bees was performed using NGS technology on an Illumina MiSeq platform in 300 PE and based on the 16S rRNA gene, regions V5-V7. Libraries were prepared according to [45] with some variations: KAPA Hi-Fi PCR Master Mix (Roche diagnostics) was used to amplify the target DNA with a maximum of 25 PCR cycles. The primers used, reported in Additional file 1: Table S1, allow the differentiation of amplicons deriving from bacteria (about 470 bp) from that deriving from plants (in pollen) plastids (about 720 bp) according to [46].
In order to exclude the amplified 720 bp band, the obtained libraries were purified with the precast E-Gel Size Select II 2% gel (Cat. Number G661012, ThermoFisher, Milan, Italy) loaded on E-Gel™ Power Snap Electrophoresis Device (ThermoFisher, Milan, Italy). Over the purify products a new PCR was performed in order to prepare the libraries with the Nextera XT DNA Library Preparation Kit (Cat. Number FC-131–2004, Illumina, Milan, Italy). The PCR products were purified in this case using magnetic beads (AMPure kit by Beckman Coulter). The purified libraries composed of 470 bp amplicons were quantified using Qubit 2.0 Fluorometer (Thermo Fisher Scientific, Milan, Italy) and the final libraries pool prepared for the sequencing at Macrogen Sequencing facilities (Seoul, South Korea). Raw sequences obtained from Macrogen were analyzed with QIIME ver. 1.9.1 [47] separating the sequencing according to the identified insect genus. R1 and R2 sequences were joined with join_paired_ends.py, allowing a minimum overlap of 5 nucleotides. Chimeras were detected with Usearch61 [48] and identified chimeras eliminated from the file. Clustering into operational taxonomic units (OTUs) (97% identity) was performed and representative OTUs were assigned using the most updated SILVA database v.136. The phylogenetic tree was generated using make_phylogeny.py (fasttree). Diversity analyses were performed with the script core_diversity_analysis.py. α–Diversity was evaluated using Chao1, Observed OTU e PD whole tree metrics; β–diversity was evaluated using both weighted and unweighted UniFrac. The obtained rarefied biom table was then used to provide information of taxonomic groups within each sample at all taxonomic levels as relative abundances. OTUs having less than 0.1% abundance were removed.
Statistics
Linear Models (LMs) have been used to compare the pathogens loads among species and among sites. The responses variables were the counts of N. bombi, N. ceranae and C. bombi. In all models, the following effects were testes: species (categorical variable: Lasioglossum, E. fervens and X. augusti), and sampling sites (categorical variable: Reserva Natural Paititi (RP), Santa Paula’s farm (SP), and Vivero Antoniucci (VA)).
To compare the gut microbiota of the different solitary bees and the effect of land use on gut microbiota composition we used univariate and multivariate analysis, respectively. To perform univariate analysis, each genus of the gut microbiota with at least 1% of relative abundance was tested for normality (Shapiro–Wilk test) and homogeneity of variance (Levene test). Depending on the assumptions of each genus, univariate comparisons were performed using ANOVA or Kruskal–Wallis followed by Tuckey test or Dunn test, respectively. Pairwise multiple comparisons were performed using Permutational multivariate analyses of variance (PERMANOVA), based on Bray–Curtis distances and 9999 permutations. All the variables included in the PERMANOVA assumed the assumption of homogeneity. A nonmetric multidimensional scaling (nMDS) based on Bray–Curtis distances and 20 minimum and 200 maximum random starts was also performed to corroborate the obtained results in PERMANOVA analysis [49]. These analyses were performed using ‘vegan’ R package version 4.2.1 [50, 51]. In order to analyze the relation between the pathogens and the gut microbiota, a Spearman correlation between each pathogen and microbiota genus was calculated. Finally, PCA analysis was performed using packages FactoMineR and factoextra, taking into consideration 21 taxa at species and/or genus level. All tests were two-tailed with a significance level of P ≤ 0.05.
Results
The taxonomical identification of solitary bees
The 56 wild bees collected in this work belonged to 3 different genera (Additional file 1: Table S2). 30 individuals were identified as Lasioglossum based on morphometric analysis with stereo microscope on the head, wings, body structure and genitals (10 individuals for each study site). The COI sequence of Lasioglossum individuals did not match with the sequences deposited the NCBI GenBank, therefore it was not possible to further discriminate at species level. However, the collected bees had the same morphological traits therefore they are belonging to the same species. A total of 11 individuals were identified as Xylocopa augusti (4 individuals from VA, 5 individuals from SP and 2 individuals from RP). Finally, 15 individuals were identified as Eucera fervens (10 individuals from SP and 5 from RP). The limited number of X. augusti and E. fervens individuals was due to the limited number of bees that was possible to recover from the collection areas at the time of sampling. In all sampling sites A. mellifera and Bombus spp. were abundantly present, although not included in the study.
The spatial characterization of the sampling sites
The 3 chosen study sites varied regarding the land use and the human impact. The RP site was considered as the area with the lowest level of human impact, considering that more than 18% of its territory is a natural reserve characterized by abundant native flora where the bees were collected. On the contrary, the area with the highest anthropic impact was SP with most of its surface (91.5%) dedicated to intensive farming. Finally, VA presents 15% of high-density residential areas, 4% of urban reserves (biological corridors), and more than 75% of intensive farming. No differences were observed regarding NVDI analysis (a) RP (NDVI mean = 0.57); (b) SP (NDVI mean = 0.49); (c) and VA (NDVI mean = 0.50). Land characterization and proportion of land use are reported in Additional file 1: Table S3.
The identified pathogens in the solitary bees gut and their spatial distribution in the sampling sites
All the collected wild bees were found uninfected by A. bombi, Ascosphaera spp., L. passim, N. apis, and AmFV when analyzed in PCR. On the contrary, the pathogens N. bombi, N. ceranae and C. bombi were detected and quantified with qPCR.
N. bombi average counts in sampled species was Log 6.57, 7.1 and 4.78 N. bombi units (NbU) for Lasioglossum, E. fervens and X. augusti, respectively Fig. 2A, Additional file 1: Fig. S1A–S1C). NbU were significantly different when comparing Lasioglossum versus E. fervens (p < 0.001), whereas the comparison of E. fervens versus X. augusti and X. augusti versus Lasioglossum, was non-significant (p = 0.453 and p = 0.198 respectively). The intensity of N. bombi varied among sites with different land uses (Fig. 2B). While the intensity of N. bombi detected SP versus VA did not show significant differences, N. bombi intensity level in RP was significantly lower when compared to SP and VA (p < 0.05).

Boxplot describing the total pathogens count regarding the solitary bee species and sampling site analyzed in this work. A qPCR results for Nosema bombi expressed in units per bee; B qPCR results for Nosema bombi expressed in units per sampling site; C qPCR results for Nosema ceranae expressed in units per bee; D qPCR results for Nosema ceranae expressed in units per sampling site; E qPCR results for Critidia bombi expressed in units per bee; F qPCR results for Critidia bombi expressed in units per sampling site; The sampling sites: VA Vivero Antoniucci; RP Reserva Paititi; SP Santa Paula Farm. * p
The level of N. ceranae was not significantly different among different bee speces and the different sampling sites (Fig. 2C–D; p > 0.05) where average counts of Log 3.32, 3.14 and 3.71 N. ceranae units (NcU) were obtained for Lasioglossum, E. fervens and X. augusti respectively (Additional file 1: Fig. S1D–F)). Nosema ceranae count, in contrast to N. bombi, did not vary within the study sites.
The average counts of C. bombi varied among wild bee species, with values of Log 5.16, Log 3.35 and Log 4.27 C. bombi units (CbU) for Lasioglossum, E. fervens and X. augusti, respectively (Fig. 2E; Additional file 1: Fig. S1G–I). CbU were significantly higher in Lasioglossum with respect to E. fervens and X. augusti (p = 0.001 and 0.043 respectively), although the comparison of X. augusti vs E. fervens was not significant (p = 0.11). Moreover, C. bombi intensity did not vary comparing SP vs VA (p > 0.05) and VA vs RP (p > 0.05), whereas significant differences were observed between SP and RP (p < 0.01; Fig. 2F) with the latter showing the lowest intensity levels of C. bombi.
A simple gut microbiome characterizes solitary bees
A total of 56 samples (30 samples for Lasioglossum, 15 samples for E. fervens and 11 for X. augusti) were subjected to NGS analysis. About 11.4 million raw reads were obtained from the sequencing (7.6 million reads for Lasioglossum, 2.33 million reads for E. fervens, 1.4 million reads for X. augusti). A total of 7 samples for Lasioglossum 4 samples of E. fervens and 2 samples of X. augusti were excluded from the downstream analysis due to the low number of reads (< 36,000). Therefore, within the 43 remained samples, a total of 5.9 million reads passed the quality control and the chimera check analysis with an average of 83 k joint reads per sample for Lasioglossum 57 k joint reads per sample for E. fervens, 52 k joint reads per sample for X. augusti. The NGS data at phylum, family and genus levels are deposited in Mendeley repository (see section “Data Availability”).
The α-diversity indexes Chao1, Observed OTUs and PD Whole Tree did not show significant variations among sampling sites (Additional file 1: Table S4). On the contrary β-diversity indexes resulted significant comparing E. fervens and X. augusti microbiota among different sites for both Weighted and Unweighted Unifrac, whereas Lassioglossum gut microbial diversity among sites was not significant (data reported in Additional file 1: Table S5).
The gut microbiome of Lasioglossum
The gut microbiome of Lasioglossum at phylum level is mainly composed of Proteobacteria (89.44%), with low percentages of Firmicutes (4.95%), Actinobacteria (1.02%), Bacteroidetes (0.48%) and Other_taxa (4.11%). Within Proteobacteria, Pseudomonadaceae and Enterobacteriaceae were the most abundant bacterial families with a relative abundance of 39.86% and 33.11%, respectively, whereas Neisseriaceae and Anaplasmataceae were present at 5.15% and 3.66%, respectively. Within the same phylum, other frequently found taxa were Rhizobiaceae (1.79%), Burkholderiaceae (1.34%), Orbaceae (1.12%), Sphingomonadaceae (0.97%), Caulobacteraceae (0.57%), and Rhodocyclaceae (0.45%). The major represented family within Firmicutes was Lactobacillaceae (4.48%) whereas whithin Actinobacteria was Bifidobacteriaceae (0.3%).
At genus level, Lasioglossum gut microbiome hosts two prevalent microbial genera: Pseudomonas and Sodalis with a relative abundance of 39.84% and 30.16%, respectively. Other prevalent taxa are Lactobacillus and Wolbachia (4.47% and 3.65%, relative abundance respectively). Notably, Snodgrassella was found in only one sample, but with a relative abundance of 69.33%. Other detected microbial genera, although present at a lower percentage, are Rhizobium (1.75%), Enterobacter (1.45%), Gilliamella (1.07%) and Sphingobium (0.76%). Finally, minor microbial taxa with a relative abundance below 1%, when grouped reach an average relative abundance of 8.13%. Prevalence values (number of colonized individuals with a target microbial taxon divided by the total analyzed individuals) results of 100%, 85.75%, 78.57 and 50% respectively for Pseudomonas, Sodalis, Wholbachia and Lactobacillus.
The gut microbiome of E. fervens
E. fervens gut microbiome is composed of 4 major phyla, Proteobacteria (72.18%), Tenericutes (15.82%), Firmicutes (3.36%) and Actinobacteria (2.11%). At family level, two major core families were detected: Pseudomonadaceae (Proteobacteria phylum) and Spiroplasmataceae (Tenericutes phylum) with 56% and 17.40% relative abundance, respectively. Less abundant, although present at relevant percentages, Enterobacteriaceae (7.19%), Orbaceae (6.12%), and Burkholderiaceae (2.48%) were detected within Proteobacteria. Lactobacillaceae families were detected with an average relative abundance of 2.40%. Other families with more than 1% relative abundance detected in E. fervens gut were Anaplasmataceae (1.66%), Neisseriaceae (1.23%) and Rhodocyclaceae (1.82%). Moreover, Microbacteriaceae belonging to the Actinobacteria phylum, were detected at 1.10%. Minor microbial taxa with a relative abundance below 1%, when grouped reached an average relative abundance of 9.81%.
In agreement with the highest taxonomy level, at genus level two main microbial genera were detected in E. ferverns: Pseudomonas, with an average relative abundance of 50.91% followed by Spiroplasma with 15.82% average relative abundance. Gilliamella, Serratia and Lactobacillus were also prevalent and show an average relative abundance of 5.41%, 2.28% and 2.18%, respectively. Other less abundant genera were Acidovorax (1.28%), Sodalis (1.17%), Weissella (0.64%), Nocardioides (0.56%) and Pantoea (0.29%). Calculated prevalence was of 100% for Pseudomonas, 45% for Spiroplasma, 54.54% for Gilliamella, 72.72% for Serratia and 63.63% for Lactobacillus.
The gut microbiome of X. augusti
The most abundant phyla present in X. augusti gut microbiome were Proteobacteria (49.01% average relative abundance), Firmicutes (35.04%), Actinobacteria (4.70%), Bacteroidetes (4.06%) and Tenericutes (1.72%). As observed for both E. fervens and Lasioglossum, Pseudomonadaceae was the most abundant bacterial family with an average relative abundance of 39.99%. However, in the case of Xylocopa, Lactobacillaceae was the second major taxa with 24.56% average relative abundance. Enterobacteriaceae, Bifidobacteriaceae and Weeksellaceae showed a relative abundance of 5.37%, 3.67% and 3.55%, respectively. Microbacteriaceae (1.11%), Anaplasmataceae (2.08%), Rhodocyclaceae (2.02%), Spiroplasmataceae (1.94%), and Streptococcaceae (2.06%) showed a low prevalence and were occasionally found with an average relative abundance above 1%. Minor microbial taxa with a relative abundance below the 1%, when grouped reached an average relative abundance of 10.11%.
In X. augusti the most abundant microbial genera were Pseudomonas and Lactobacillus with an average relative abundance of 36.05% and 30.56%, respectively and a prevalence of 100% and 88.89% respectively. These genera were detected in the majority of X. augusti samples, showing a high frequency of colonization. Less abundant taxa although frequently found were Apibacter and Bifidobacterium that accounted for an average amount of 3.05% and 3.17%, respectively, and with a prevalence of 44.45% and 55.55% respectively. The genus Wolbachia (average 1.88%), Spiroplasma (1.49%), Serratia (1.69%), Lactococcus (1.48%), Pantoea (1.33%) and Methyloversatilis (1.08%) are occasionally found with a relative abundance above 1%. Finally, a very large number of microbial taxa with a relative abundance lower than 1% were found in the X. augusti gut, grouped in “other” with a total relative abundance of 11.6%.
The comparison of the different microbiome profiles within solitary bees shows shared microbial taxa and some peculiar genera
In all tested solitary bees the most represented and co-shared genus was Pseudomonas, however several differences were observed in pairwise comparisons. The p values obtained from these comparisons are summarized in Table 1. The genera Acidovorax, Geothermobacter, Gilliamella, Methyloversatilis, Nocardioides, Panotea, Serratia, Spiroplasma and the group “Other” were significantly more abundant in E. fervens compared to Lasioglossum (p<0.05). Bifidobacterium, Brevundimonas, Chryseobacterium, Massilia, Rhizobium and Sphingobium abundances were higher in Lasioglossum compared to the other bees (p= 0.001).
Significant differences were found comparing gut bacteria relative abundance of E. fervens versus X. augusti. Apibacter, Bifidobacterium, Lactococcus and Spiroplasma were significantly higher in X. augusti (p < 0.05) whereas Lactobacillus, Nocardioides and Snodgrassella showed higher relative abundance in E. fervens (p < 0.05). The remaining bacterial genera did not present significant differences. Comparing the gut microbial groups relative abundance between X. augusti and Lasioglossum, the genera Apibacter, Bifidobacterium, Geothermobacter, Lactobacillus, Lactococcus, Panotea, Spiroplasma and Weissella, presented significant higher relative abundance in X. augusti (all p < 0.05). On the contrary, Brevundimonas, Chryseobacterium, Massilia, Rhizobium, Snodgrassella, Sodalis and Sphingobium genera showed significant higher relative abundance in Lasioglossum (all p < 0.05). Finally, PERMANOVA analysis (F = 6.20, p < 0.001) revealed significant differences for all pairwise comparisons (all p < 0.01). The NMDS analysis presented good non-metric and linear fit to the observed dissimilarity, and a fair stress level (Fig. 3A) and showed a relative segregation between species, with an area of overlap close to the origin of the axes (p < 0.05, Fig. 3B).

A–C Gut bacteria assemblage NMDS. Non-metric multi-dimensional scaling (NMDS) for the gut bacteria assemblage. Each points represent individuals. A Non-metric and linear fit (R2) and stress level between the ordination distance and the observed dissimilarity, indicating the analyses were accurate. B The bacterial assemblage varied within species C but no differences were observed in the bacterial assemblage at locality scale
The land use partially affects the gut microbiome of the solitary bees
The 22 most abundant microbial genera populating the gut microbiome of the three solitary bees were considered for the correlation analysis with the environment. Among these genera, 20 did not show any statistical difference among the different study sites in all solitary bees (p > 0.05). On the contrary Snodgrassella and Nocardioides recorded a higher relative abundance in RP when compared to the VA (p < 0.001 and p < 0.05, respectively). Also, while both genera were equally present in SP and VA sampling sites (p = 1.000 and p = 0.514, respectively), Snodgrasella showed higher abundance in RP if compared to SP (p = 0.011). This difference was not observed for Nocardioides (p = 0.304). Regarding the comparisons of each bacterial taxon among the analyzed species, two genera displayed a similar relative abundance in all pollinator species: Pseudomonas (p = 0.306) and Wolbachia (p = 0.491). PERMANOVA analysis showed differences between land uses (F = 2.02, p < 0.05), however, in pairwise comparisons, after Bonferroni correction, no effect of the different land uses was observed on gut microbiome composition (p > 0.05). The NMDS analysis showed a high overlap among sampling sites (Fig. 3C). Even when the Bonferroni correction shows that there is no significant effect of the land use over the gut microbiome, the preliminar obtanined differences of the PERMANOVA reflects a partial tendency illustrated on Fig. 3C where the size of the circle is positively correlated with diversity of the bacterial genus.
The bee pathogens are shaping the gut microbiome of solitary bees
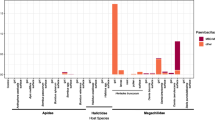
The three pathogens detected showed different levels of correlation with the microbial taxa populating the gut microbiome of the analyzed solitary bees (Fig. 4). While N. ceranae did not show relevant correlation with the gut microbial taxa described (p > 0.05), N. bombi and C. bombi strongly influenced the microbiome profile positively or negatively correlating with multiple microbial taxa. On the one hand, N. bombi was negatively correlated with Bifidobacterium, Apibacter and Lactococcus (p < 0.05), whereas it was positively correlated with Snodgrassella and Nocardioides (p < 0.05). C. bombi positively correlated with Rizobium, Sphingobium, Massilia, Brevundimonas (p < 0.01) and Sodalis (p < 0.05) whereas it negatively correlated with Pantoea, Spiroplasma, Serratia, Acidovorax and other minor microbial taxa (p < 0.01) (Fig. 4).

Spearman heat-map for pathogens and intestinal bacteria correlation. Heat-map for Spearman's correlation indices. Positive values (in red scales) represent positive correlation between bacterial genera and pathogen, while negative values (in blue scale) represent a negative correlation. * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001
An integrate effect of pathogens, land use and bee species over the gut microbiome
As a resume, the Principal Component Analysis (PCA) analysis showed the synergy between the land use, pathogens, and bee species (Fig. 5). Pathogens were found as the drivers of the gut microbiome shaping, especially affecting abundance of minor microbial groups in the gut microbiome and in the different sampling sites.

PCA analysis. A PCA was performed with 21 taxa at genus level; confidence ellipses are shown in the graph. The graph includes the top ten variables with the highest contribution. RP: Reserva Natural Paititi; SP: Santa Paula; VA: Vivero Antoniucci. B PCA considering E. fervens and the sampling sites RP and SP. C PCA considering Lasioglossum spp., and the sampling sites RP, SP and VA. D PCA considering X. augusti, and the sampling sites SP and VA
Discussion
In this study the gut microbiome of three important solitary bees of South America (X. augusti, E. fervens and Lasioglossum) was studied, also examining the presence and load of potential gut pathogens and evaluating the effect of land use on microbiota and pathogen composition.
Pathogen distribution in solitary bees
This research showed a wide and homogeneous spread of N. ceranae in all the wild pollinators sampled. The role of A. mellifera as a vector of a relevant number of pathogens, including N. ceranae, to wild bees has been described by Graystock et al. [52] and Furst et al. [53]. Since A. mellifera was recorded in all sampling sites with a high abundance, this could explain the spread of N. ceranae spores to flowers and consequently to local pollinators [54]. The homogeneity of N. ceranae on the wild pollinators sampled in the different territories is not surprising, also considering that N. ceranae spillover was defined as a new pandemic in pollinators [55]. Concerning N. bombi, this work reported its presence in all the studied bee species. Only few data are available on N. bombi spillover from the social bee Bombus spp. to other wild solitary bees. Differently from our results, the only other work investigating the N. bombi spread from Bombus to Xylocopa spp. did not detect its presence in X. augusti in Chile [56], probably due to the recent introduction of this solitary bee in Chile. The pathogen C. bombi seems to be affected by both the land use and the bee species. A similar result was found for N. ceranae in the study of Theodorou et al. [34], which showed that land use changes affected the nutritional resources correlated with the pathogen. In the Natural Reserve Paititi the recorded presence of Bombus spp. was twice when compared with the other sampling areas (data not shown in this manuscript). Bombus pauloensis is the most widespread native bee of the Buenos Aires province [57], and the ideal carrier of C. bombi, thus explaining the higher abundance of C. bombi in X. augusti sampled in the same area. Even when flowers could act as potential vectors for C. bombi, this occasionally happened with E. fervens since it is a highly specialized pollinator of pumpkins (e.g. Cucurbita maxima) [58]. It has already been highlighted that nutrition diversity impacts the load of C. bombi and the response of the solitary bees to this pathogen [59]. This may explain why E. fervens, having a diversified diet with respect to other solitary bees, has a lower load of C. bombi in comparison to Lasioglossum that feeds on a more diversified floral resources [60].
The peculiar gut microbiome of solitary bees
The gut microbiome of honeybees (Apis mellifera) was one of the first insect microbiome studied with NGS approaches, and it was proposed as a model for microbiome research [61]. The great attention and interest raised by the honeybee microbiome has led to a detailed knowledge on its acquirement, diversity, shaping by external factors [45, 62,63,64], and on its function in relation to the brain-gut axis [42, 65]. On the other hand, less is known for other pollinators and especially for solitary bees probably because of the difficulties in collection and study. The few studies available on non-social bee species showed a more variable and less distinctive microbiome if compared to social bees [66,67,68]. Solitary bees are also usually prone to maternally transmitted bacteria [69] and the acquisition of a homogeneous gut microbiome is presumably harder, therefore complicating the determination of the core taxa inhabiting the gut. An interesting debate on the definition of core taxa is still ongoing: Ainsworth et al. [70] described the core microbiome as the microbial taxa that occur with a frequency between 30 and 95% of relative abundance. Risely [71], on the other hand, gave less importance on the intensity factor and rather considered prevalence as the most important factor. For instance, in honey bees, the genus Bifidobacterium is considered a core microbial taxa despite being often found at low relative abundance (2–5%). For this reason, in the attempt to describe the core microbiota of the analyzed solitary bees, the abundance of Bifidobacterium in honey bees was taken as a reference threshold, in this study we focused our attention on the microbial genera with an equivalent or higher prevalence.
In this study we sampled Lasioglossum solitary bees, although it is known that some species of Lasioglossum show an incipient of social behavior. The two major microbial groups in the gut microbiome of Lasioglossum were Lactobacillus and Sodalis in accordance with Mayr et al., [72] and Rubin et al., [73]. Lactobacillaceae is an important and frequently found family in solitary and social bees responsible for the biosynthesis of some vitamins (e.g.: riboflavin and thiamin), amino acids [38] and short chain fatty acids, positively impacting social behavior and learning of the bees [65]. The majority of the NGS reads could be taxonomically assigned at genus level but not at species level, and possibly Lasioglossum is a reservoir of novel Lactobacillus species different from those isolated from Apis and Bombus spp. Sodalis is a microbial taxon widely present in the gut of solitary bees like Megachile rotundata [74], Osmia spp. and Lasioglossum [69] rather than social bees where it is present in relative low proportions [73]. Sodalis is a maternally transmitted microbial taxon in solitary bees [69] and this taxon showed differences in abundances among Lasioglossum species with different social behaviors, with higher abundance in species with a lower social behavior [73]. According to these findings, a very high relative abundance was found in the solitary Lasioglossum studied in this work. Sodalis might be an obligate endosymbiont for Lasioglossum as it is for other insects [75]. While other symbionts like Gilliamella apicola and Snodgrassella alvi have shown a negative correlation with C. bombi [76], our results demonstrated a Sodalis positive correlation with this Trypanosomatidae. Similar correlations between Sodalis and other Trypanosomatids like mutualistic Trypanosoma brucei rhodesiense have been reported in the literature for the Tse-tse fly Glossina spp. [77].
In Lasioglossum the gut microbiome also showed a strong prevalence of Pseudomonas and Wolbachia. The co-occurrence of these two genera was also described in other insect’s gut microbiome like the cricket Gryllus veletis [78], although Pseudomonas was considered as a probable pathogen that varied its proportion with seasonality and according to Wolbachia´s abundance. Interestingly, Ge et al., [79] found similar proportion of Pseudomonas and Wolbachia in the reproductive system of Paederus fuscipes (coleoptera), where Pseudomonas was found to contribute to the defense from predators thanks to the production of a toxic compound (Pederin), without decreasing the performance of the hosts. Moreover, Wolbachia was found to cope with adverse conditions triggered by Pseudomonas. The role of Wolbachia in the defense of the host from viral and bacterial pathogens is a recognized trait in different classes of insects [80, 81].
E. fervens gut microbiota has never been described despite its well-recognized importance in the pollination of some commercial crops in South America. Here, a strong presence of Pseudomonas was detected as occurred with Lasioglossum and X. augusti, and this is discussed below. Spiroplasma, the second most abundant taxa in E. fervens, is a widely found bacterium in ground beetles like Pseudophonus [82]. Spiroplasma species have been shown to protect the host by increasing defenses against pathogens [83] and it directly interacts with secreted antimicrobial molecules [84]. In fact, in this work Spiroplasma presence negatively correlated with C. bombi load, confirming this genus as a potential enhancer of the defense capabilities of the gut microbiome. Finally, Gilliamella and Lactobacillus were also detected with a high relative abundance. In honey bees, Gilliamella is the main responsible for pollen degradation together with Lactobacillus. In fact, the Gilliamella genus is reported to possess a complete metabolic pathway for pectin and hemicellulose degradation [38, 61]. E. fervens is well known to nourish mainly on pumpkin pollen [85], therefore this rich diet may justify the presence of Gilliamella. To the best of our knowledge, just one report on the gut microbiome of an Eucera species, different from E. fervens, was published to date [86]. Shapiro et al. [86] investigated the gut microbiome at order level of Eucera pruinosa and showed a major presence of Lactobacillales, Pseumonadales and Cytophagales that broadly reflects our findings.
The latest reports on the Xylocopa spp. gut microbiome vary considerably with Xylocopa species and the different sampling sites. Holley et al. [87], found a very high proportion of Bombiscardovia, Pseudomonas, Xenorhabdus, and different Lactobocillaceae genera (Apilactobacillus, Bombilactobacillus, Lactobacillus and Latilactobacillus) in Xylocopa micans and Xylocopa tabaniformis. Differently, Handy et al. [88], studied Xylocopa sonorina and Xylocopa tabaniformis sampled from California and Arizona detected Apibacter, Schmidhempelia, Enteromonas, Enterobacter and Fructobacillus as the main genera. The results shown by Handy et al. [88], supported the influence of the landscape on the gut microbiome. Our results did not support the influence of land use on the gut microbiome of X. augusti, nevertheless the differences in the gut microbiome profiles obtained in this work, when compared to Handy et al. [88] and Holley et al. [87], could be explained both by the different land use and species considered. However, in our case, within Lactobacillaceae, only the genus Lactobacillus was detected, differently from Holley et al. [87]. In our study, Apilactobacillus, Bobilactobacillus and Latilactobacillus were below the limit of detection, perhaps due to the impact of land use on the gut microbiome. This is supported by the fact that a relatively low proportion of Lactobacilaceae was also detected in Lasioglossum and E. fervens in the same conditions and sampling period. Pseudomonas was frequently found in the Xylocopa individuals sampled by Holley et al. [88] in Texas but not in Handy et al. [88] samples obtained in California and Arizona. This confirms that gut microbiome acquisition is strongly dependent on the environmental conditions.
Pseudomonas as the major group in solitary bees
The role of Pseudomonas in the gut microbial communities of the solitary bees analyzed in this study is not well understood yet, but in social bees the presence of Pseudomonas seems to be correlated with the presence of molecules of anthropic origin such as the antibiotic tylosin [45] or more probably to bees collecting water where Pseudomonas is widely diffused [89]. A possible explanation of the high Pseudomonas abundance in the analyzed bees within this study may be the extensive use of glyphosate in the area from which bees were sampled. In the last three decades, in the Pampas region, the use of glyphosate has increased because of the spread of glyphosate resistant crops [90]. It is well known that Pseudomonas can catabolize this molecule and use it as additional carbon source [91,92,93] and, therefore, its abundance may be an adaptation to contaminated nectar. Indeed, a remarkable amount of honey samples resulted to be contaminated with glyphosate worldwide [94, 95] but also in the sampling district within the Pampas of this work [96]. Recently, Motta et al., [96] showed that glyphosate can perturb the gut microbiome of honey bees, but the perturbation might also be an adaptation to the xenobiotics. To confirm the role of insect gut bacteria in xenobiotic degradation, it has been observed that in the wasp Nasonia vitripennis both the gut bacteria Serratia and Pseudomonas contributed to atrazine degradation, conferring resistance to wasp populations [93].
Conclusion
This work supports the hypothesis that land use and anthropization pressure contribute to weakening bee health predisposing bees to pathogens proliferation. Anthropization itself contributes to biotic stressors spread and to biotic and abiotic stressors incidence, which can impact on the microbiota composition. Although there was a trend towards significant variation in the gut microbial composition of native bees, our results did not show a direct correlation between land use and gut microbiota changes in solitary bees. However, land use influenced the presence and load of pathogens, which are the main contributors to the shaping of the gut microbiota. Therefore, the microbiota changes can be an indirect effect of land use caused by decreased nutrient sources and water availability, and by the human mediated spread of social pollinators. Additionally, this study characterized for the first time the core microbiome of wild pollinators of Argentina, contributing to the knowledge on the solitary bee microbiome composition. Nevertheless, the current knowledge does not allow a clear understanding whether the microbiome changes can positively or negatively influence the host growth and health, and consequently the life span of studied solitary pollinators.
Availability of data and materials
NGS raw sequence data have been submitted to NCBI repository under the Sequence Read Archive (SRA) databases under the Bioproject N° PRJNA799463. Accession numbers SAMN25173657—SAMN25173686 belong to Lasioglossum samples; SAMN25173642—SAMN25173656 belong to Eucera fervens samples; SAMN25173688—SAMN25173697 belong to Xylocopa augusti samples. The datasets generated and/or analyzed during the current study are available in the Mendelay Data repository: Published: 12 December 2022 |Version 1| https://doi.org/10.17632/jrs7rr8vw5.1.
References
Gallai N, Salles JM, Settele J, Vaissière BE. Economic valuation of the vulnerability of world agriculture confronted with pollinator decline. Ecol Econ. 2009;68:810–21.
Michener C. The bees of the world. 2nd ed. Baltimore and London: Johns Hopkins University Press; 2007.
Ollerton J, Johnson SD, Hingston AB. Geographical variation in diversity and specificity of pollination systems. In: Waser NM, Ollerton J, editors. Plant-pollinator interactions: from specialization to generalization. Chicago: The University of Chicago Press; 2006. p. 283–308.
Javorek SK. Comparative pollination effectiveness among bees (Hymenoptera: Apoidea) at lowbush blueberry (Ericacea: Vaccinium angustifolium Ait.). Ann Entomol Soc Am. 2002;95:345–51.
Kremen C, Williams NM, Thorp RW. Crop pollination from native bees at risk from agricultural intensification. Proc Natl Acad Sci. 2002;99:16812–6.
Klein AM, Steffan-Dewenter I, Tscharntke T. Pollination of Coffea canephora in relation to local and regional agroforestry management. J Appl Ecol. 2003;40:837–45.
Winfree R. The conservation and restoration of wild bees. Ann NY Acad Sci. 2010;1195(1):169–97.
Garibaldi LA, Carvalheiro LG, Leonhardt SD, Aizen MA, Blaauw BR, Isaacs R, Kuhlmann M, Kleijn D, Klein AM, Kremen C, Morandin L, Scheper J, Winfree R. From research to action: enhancing crop yield through wild pollinators. Front Ecol Environ. 2014;12(8):439–47.
Lopresti MF, Di Bella CM, Degioanni AJ. Relationship between MODIS-NDVI data and wheat yield: a case study in Northern Buenos Aires province. Argent Inf Process Agric. 2015;2(2):73–84.
Ramello PJ, Álvarez LJ, Almada V, Lucia M. The melittofauna and its floral associations in a natural riparian forest in Buenos Aires province. Argent J Apicult Res. 2021;60(2):241–54.
Lucia M, Alvarez LJ, Abrahamovich AH. Large carpenter bees in Argentina: systematics and notes on the biology of Xylocopa subgenus Neoxylocopa (Hymenoptera: Apidae). Zootaxa. 2014;3754:201–38.
Montalva JM, Allendes JL, Lucia M. The large carpenter bee Xylocopa augusti (Hymenoptera: Apidae): new record for Chile. J Melittol. 2013;12:1–6.
Lucia M, Tellería MC, Ramello PJ, Abrahamovich AH. Nesting ecology and floral resource of Xylocopa augusti Lepeletier de Saint Fargeau (Hymenoptera, Apidae) in Argentina. Agric Forest Entomol. 2017;19
Keasar T. Large carpenter bees as agricultural pollinators. Psyche. 2010;1–7.
Kocher SD, Li C, Yang W, Tan H, Yi SV, Yang X, Hoekstra HE, Zhang G, Pierce NE, Yu DW. The draft genome of a socially polymorphic halictid bee Lasioglossum Albipes. Genome Biol. 2013;14(12):1–14.
González-Vaquero R, Gravel AI, Devoto M. Information retrieved from specimens at Natural History Collections can improve the quality of field-based ecological networks. Commun Ecol. 2014;15(2):187–93.
Engel MS. Classification of the bee tribe Augochlorini (Hymenoptera: Halictidae). B Am Mus Nat Hist. 2000;2000(250):1–89.
Packer L, Gibbs J, Sheffield C, Hanner R. DNA barcoding and the mediocrity of morphology. Mol Ecol Resour. 2009;9:42–50.
González-Vaquero RA. Revisión sistemática del género Halictillus (Hymenoptera: Halictidae: Augochlorini) en la Argentina. Rev Soc Entomol Argent. 2010;69(1–2):65–89.
Fernandez de Landa G, Meroi Arcerito FR, Corti C, Revainera PD, Nicolli AR, Zumpano F, Brasesco C, Quintana S, Fernandez de Landa M, Ramos F, Petrigh R, Eguaras MJ, Galetto L, Maggi M. Can the exotic pathogen Nosema ceranae affect the amount of Cucurbita maxima pollen grains transported by the native bee Eucera fervens? Arthropod-Plant Int. 2022;1–9.
Dorchin A, López-Uribe MM, Praz CJ, Griswold T, Danforth BN. Phylogeny, new generic-level classification, and historical biogeography of the Eucera complex (Hymenoptera: Apidae). Mol Phylogenet Evol. 2018;119:81–92.
Hurd JRPD, Linsley EG. South American squash and gourd bees of the genus Peponapis (Hymenoptera: Apoidea). Ann Entomol Soc Am. 1967;60(3):647–61.
Hurd PD, Linsley EG, Whitaker TW. Squash and gourd bees (Peponapis, Xenoglossa) and the origin of the cultivated Cucurbita. Evolution. 1971;25(1):218–34.
Krug C, Alves-dos-Santos I, Cane J. Visiting bees of Cucurbita flowers (Cucurbitaceae) with emphasis on the presence of Eucera fervens Smith (Eucerini-Apidae)–Santa Catarina, southern Brazil. Oecol Aust. 2010;14(1):128–39.
Vray S, Rollin O, Rasmont P, Dufrêne M, Michez D, Dendoncker N. A century of local changes in bumblebee communities and landscape composition in Belgium. J Insect Conserv. 2019;23(3):489–501.
Dicks LV, Breeze TD, Ngo HT, Senapathi D, An J, Aizen MA, Basu P, Buchori D, Galetto L, Garibaldi LA, Gemmill-Herren B, Howlett BG, Imperatriz-Fonseca VL, Johnson SD, Kovács-Hostyánszki A, Kwon YJ, Lattorff HMG, Lungharwo T, Seymour CL, Vanbergen AJ, Potts SG. A global-scale expert assessment of drivers and risks associated with pollinator decline. Nat Ecol Evol. 2021;5(10):1453–61.
Brown MJF, Paxton RJ. The conservation of bees: a global perspective. Apidologie. 2009;40:410–6.
Maggi M, Antúnez K, Invernizzi C, Aldea P, Vargas M, Negri P, Brasesco C, De Jong D, Message D, Teixeira EW, Principal J, Barrios C, Ruffinengo S, Da Silva RR, Eguaras M. Honeybee health in South America. Apidologie. 2016;47(6):835–54.
Maggi M, Quintana S, Revainera PD, Porrini LP, Meroi Arcerito FR, Fernández de Landa G, Brasesco C, Di Gerónimo V, Ruffinengo SR, Eguaras MJ. Biotic stressors affecting key apiaries in Argentina. Bee World. 2020;97(2):45–52.
Goulson D, Nicholls E, Botías C, Rotheray EL. Bee declines driven by combined stress from parasites, pesticides, and lack of flowers. Science. 2015;347(6229):1255957.
Rutrecht ST, Brown MJ. The life-history impact and implications of multiple parasites for bumble bee queens. Int J Parasitol. 2008;38(7):799–808.
Meeus I, Pisman M, Smagghe G, Piot N. Interaction effects of different drivers of wild bee decline and their influence on host–pathogen dynamics. Curr Opin Insect Sci. 2018;26:136–41.
Revainera PD, Quintana S, Fernández de Landa G, Meroi Arcerito F, Lucía M, Abrahamovich AH, Plischuk S, Eguaras MJ, Maggi MD. Phoretic mites on South American bumblebees (Bombus spp.) as parasite carriers: a historical input. Apidologie. 2020;51(4):455–64.
Theodorou P, Radzevičiūtė R, Settele J, Schweiger O, Murray TE, Paxton RJ. Pollination services enhanced with urbanization despite increasing pollinator parasitism. P Roy Soc B-Biol Sci. 1833;2016(283):20160561.
Dillon RJ, Dillon VM. The gut bacteria of insects: nonpathogenic interactions. Annu Rev Entomol. 2004;49(1):71–92.
Martinson VG, Moy J, Moran NA. Establishment of characteristic gut bacteria during development of the honeybee worker. Appl Environ Microb. 2012;78(8):2830–40.
Alberoni D, Gaggìa F, Baffoni L, Di Gioia D. Beneficial microorganisms for honey bees: problems and progresses. Appl Microbiol Biot. 2016;100(22):9469–82.
Alberoni D, Di Gioia D, Baffoni L. Alterations in the microbiota of caged honeybees in the presence of Nosema ceranae infection and related changes in functionality. Microb Ecol. 2022;1–16.
Itoh H, Tago K, Hayatsu M, Kikuchi Y. Detoxifying symbiosis: microbe-mediated detoxification of phytotoxins and pesticides in insects. Nat Prod Rep. 2018;35(5):434–54.
Raymann K, Moran NA. The role of the gut microbiome in health and disease of adult honey bee workers. Curr Opin Insect Sci. 2018;26:97–104.
Hubert J, Bicianova M, Ledvinka O, Kamler M, Lester PJ, Nesvorna M, Kopecky J, Erban T. Changes in the bacteriome of honey bees associated with the parasite Varroa destructor, and pathogens Nosema and Lotmaria passim. Microb Ecol. 2017;73(3):685–98.
Jones JC, Fruciano C, Hildebrand F, Al Toufalilia H, Balfour NJ, Bork P, Engel P, Ratnieks FLW, Hughes WOH. Gut microbiota composition is associated with environmental landscape in honey bees. Ecol Evol. 2018;8(1):441–51.
Carlson TN, Ripley DA. On the relation between NDVI, fractional vegetation cover, and leaf area index. Remote Sens Environ. 1997;62(3):241–52.
Garrido PM, Porrini MP, Alberoni D, Baffoni L, Scott D, Mifsud D, Eguaras M, Di Gioia, D. Beneficial bacteria and plant extracts promote honey bee health and reduce nosema ceranae infection. Probiotics Antimicrob. (2023);1–16.
Alberoni D, Favaro R, Baffoni L, Angeli S, Di Gioia D. Neonicotinoids in the agroecosystem: in-field long-term assessment on honeybee colony strength and microbiome. Sci Total Environ. 2021;762:144116.
Hanshew AS, Mason CJ, Raffa KF, Currie CR. Minimization of chloroplast contamination in 16S rRNA gene pyrosequencing of insect herbivore bacterial communities. J Microbiol Meth. 2013;95(2):149–55.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Knight R. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–6.
Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G, Ciulla D, Tabbaa D, Highlander SK, Sodergren E, Methé B, DeSantis TZ, The Human Microbiome Consortium, Petrosino JF, Knight R, Birren BW. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011;21(3):494–504.
Palacio FX, Apodaca MJ, Crisci JV. Análisis multivariado para datos biológicos: teoría y su aplicación utilizando el lenguaje R. Fundación de Historia Natural Félix de Azara Ed; 2020.
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. 2022. URL https://www.R-project.org/
Oksanen J, Simpson GL, Blanchet FG, Kindt R, Legendre P, Minchin PR, Weedon J. vegan: community ecology package. R package ver. 2.6–2. 2022.
Graystock P, Goulson D, Hughes WO. The relationship between managed bees and the prevalence of parasites in bumblebees. PeerJ. 2014;2:e522.
Fürst MA, McMahon DP, Osborne JL, Paxton RJ, Brown MJF. Disease associations between honeybees and bumblebees as a threat to wild pollinators. Nature. 2014;506(7488):364–6.
Graystock P, Goulson D, Hughes WO. Parasites in bloom: flowers aid dispersal and transmission of pollinator parasites within and between bee species. P Roy Soc B-Biol Sci. 1813;2015(282):20151371.
Grupe AC, Quandt CA. A growing pandemic: a review of Nosema parasites in globally distributed domesticated and native bees. PLoS Pathog. 2020;16(6):e1008580.
Fernández K, Alcaíno J, Sepúlveda D, Medel R. Assessment of intestinal parasites in the coexisting Bombus terrestris (Apidae) and Xylocopa augusti (Apidae) in central Chile. Rev Chil Hist Nat. 2020;93.
Abrahamovich A, Díaz N, Lucia M. Identificación de las “abejas sociales” del género Bombus (Hymenoptera, Apidae) presentes en la Argentina: clave pictórica, diagnosis, distribución geográfica y asociaciones florales. Rev Fac Agron La Plata. 2007;106(2):165–76.
Petersen JD, Reiners S, Nault BA. Pollination services provided by bees in pumpkin fields supplemented with either Apis mellifera or Bombus impatiens or not supplemented. PLoS ONE. 2013;8(7):e69819.
Figueroa LL, Grincavitch C, McArt SH. Crithidia bombi can infect two solitary bee species while host survivorship depends on diet. Parasitology. 2021;148(4):435–42.
Aguilar Sierra CI, Smith Pardo AH. Abejas visitantes de Mimosa pigra L. (Mimosaceae): comportamiento de pecoreo y cargas polínicas. Acta Biol Colomb. 2009;14(1):109–20.
Zheng H, Steele MI, Leonard SP, Motta EV, Moran NA. Honey bees as models for gut microbiota research. Lab Anim. 2018;47(11):317–25.
Baffoni L, Alberoni D, Gaggìa F, Braglia C, Stanton C, Ross PR, Di Gioia D. Honeybee exposure to veterinary drugs: how is the gut microbiota affected? Microbiol Spect. 2021;9(1):e00176-e221.
Motta EV, Raymann K, Moran NA. Glyphosate perturbs the gut microbiota of honey bees. P Natl Acad Sci USA. 2018;115(41):10305–10.
Almeida EL, Ribiere C, Frei W, Kenny D, Coffey MF, O’Toole PW. Geographical and seasonal analysis of the honeybee microbiome. Microb Ecol. 2023;85:765–78.
Zhang Z, Mu X, Shi Y, Zheng H. Distinct roles of honeybee gut bacteria on host metabolism and neurological processes. Microbiol Spectr. 2022;10(2):e02438-e2521.
McFrederick QS, Rehan SM. Wild bee pollen usage and microbial communities co-vary across landscapes. Microb Ecol. 2019;77(2):513–22.
Cohen H, McFrederick QS, Philpott SM. Environment shapes the microbiome of the blue orchard bee. Osmia Lignaria Microb Ecol. 2020;80(4):897–907.
Subta P, Yodsuwan P, Yongsawas R, In-On A, Warrit N, Panha S, Khongphinitbunjong K, Chantawannakul P, Attasopa K, Disayathanoowat T. Bacterial communities in three parts of intestinal tracts of carpenter bees (Xylocopa tenuiscapa). Insects. 2020;11(8):497.
Saeed A, White JA. Surveys for maternally-inherited endosymbionts reveal novel and variable infections within solitary bee species. J Invertebr Pathol. 2015;132:111–4.
Ainsworth TD, Tracy D, Krause L, Bridge T, Torda G, Raina JB, Zakrzewski M, Gates RD, Ruth DG, Padilla-Gamiño JL, Spalding HL, Smith C, Woolsey ES, Bourne DG, Bongaerts P, Hoegh-Guldberg O, Leggat W. The coral core microbiome identifies rare bacterial taxa as ubiquitous endosymbionts. ISME J. 2015;9(10):2261.
Risely A. Applying the core microbiome to understand host–microbe systems. J Anim Ecol. 2020;89(7):1549–58.
Mayr AV, Keller A, Peters MK, Grimmer G, Krischke B, Geyer M, Schmitt T, Steffan-Dewenter I. Cryptic species and hidden ecological interactions of halictine bees along an elevational gradient. Ecol Evol. 2021;11(12):7700–12.
Rubin BE, Sanders JG, Turner KM, Pierce NE, Kocher SD. Social behaviour in bees influences the abundance of Sodalis (Enterobacteriaceae) symbionts. Roy Soc Open Sci. 2018;5(7):180369.
McFrederick QS, Wcislo WT, Hout MC, Mueller UG. Host species and developmental stage, but not host social structure, affects bacterial community structure in socially polymorphic bees. FEMS Microbiol Ecol. 2014;88(2):398–406.
Fukatsu T, Koga R, Smith WA, Tanaka K, Nikoh N, Sasaki-Fukatsu K, Yoshizawa K, Dale C, Clayton DH. Bacterial endosymbiont of the slender pigeon louse, Columbicola columbae, allied to endosymbionts of grain weevils and tsetse flies. Appl Environ Microbiol. 2007;73(20):6660–8.
Kwong WK, Moran NA. Cultivation and characterization of the gut symbionts of honey bees and bumble bees: description of Snodgrassella alvi gen. nov., sp. nov., a member of the family Neisseriaceae of the Betaproteobacteria, and Gilliamella apicola gen. nov., sp. nov., a member of Orbaceae fam. nov., Orbales ord. nov., a sister taxon to the order ‘Enterobacteriales’ of the Gammaproteobacteria. Int J Syst Evol Microbiol. 2013;63(6):2008–18.
Dale C, Maudlin I. Sodalis gen. nov. and Sodalis glossinidius sp. nov., a microaerophilic secondary endosymbiont of the tsetse fly Glossina morsitans morsitans. Int J Syst Evol Microbiol. 1999;49(1):267–75.
Ferguson LV, Dhakal P, Lebenzon JE, Heinrichs DE, Bucking C, Sinclair BJ. Seasonal shifts in the insect gut microbiome are concurrent with changes in cold tolerance and immunity. Funct Ecol. 2018;32(10):2357–68.
Ge C, Hu J, Zhao Z, Hoffmann AA, Ma S, Shen L, Fang J, Zhu J, Yu W, Jiang W. Phylogeny and density dynamics of Wolbachia infection of the health pest Paederus fuscipes Curtis (Coleoptera: Staphylinidae). Insects. 2020;11(9):625.
Wong ZS, Hedges LM, Brownlie JC, Johnson KN. Wolbachia-mediated antibacterial protection and immune gene regulation in Drosophila. PLoS ONE. 2011;6(9):e25430.
Balaji S, Deepthi KNG, Prabagaran SR. Native Wolbachia influence bacterial composition in the major vector mosquito Aedes aegypti. Arch Microbiol. 2021;203(8):5225–40.
Magagnoli S, Alberoni D, Baffoni L, Martini A, Marini F, Di Gioia D, Mazzon M, Marzadori C, Campanelli G, Burgio G. The ground beetle Pseudoophonus rufipes gut microbiome is influenced by the farm management system. Sci Rep. 2022;12(1):22638.
Engel P, Moran NA. Functional and evolutionary insights into the simple yet specific gut microbiota of the honey bee from metagenomic analysis. Gut Microbes. 2013;4(1):60–5.
Ballinger MJ, Perlman SJ. The defensive Spiroplasma. Curr Opi Insect Sci. 2019;32:36–41.
Thorp RW. Structural, behavioral, and physiological adaptations of bees (Apoidea) for collecting pollen. Ann Mo Bot Gard. 1979;788–812.
Shapiro LR, Youngblom M, Scully ED, Rocha J, Paulson JN, Klepac-Ceraj V, Cibrián-Jaramillo A, López-Uribe MM. Bacterial communities of herbivores and pollinators that have co-evolved Cucurbita spp. BioRxiv. 2019;691378.
Holley JAC, Jackson MN, Pham AT, Hatcher SC, Moran NA. Carpenter bees (Xylocopa) harbor a distinctive gut microbiome related to that of honey bees and bumble bees. Appl Environ Microbiol. 2022;88(13):e00203-e222.
Handy M, Sbardellati D, Yu M, Saleh N, Ostwald M, Vannette R. Incipiently social carpenter bees (Xylocopa) host distinctive gut bacterial communities and display geographic structure as revealed by full-length PacBio 16S rRNA sequencing. Mol Ecol. 2023;32(6):1530–43.
D’alvise P, Böhme F, Codrea MC, Seitz A, Nahnsen S, Binzer M, Rosenkranz P, Hasselmann M. The impact of winter feed type on intestinal microbiota and parasites in honey bees. Apidologie. 2018;49(2):252–64.
Galetto L, Aizen MA, Arizmendi MDC, Freitas BM, Garibaldi LA, Giannini TC, Lopes AV, Ariadna V, Do Espírito Santo MM, Maués MM, Nates Parra GPG, Rodríguez JI, Quezada Euán JJG, Vandame R, Viana BF, Imperatriz Fonseca VL. Risks and opportunities associated with pollinators’ conservation and management of pollination services in Latin America. Ecol Aust. 2022;32:55–76.
Zhao H, Tao K, Zhu J, Liu S, Gao H, Zhou X. Bioremediation potential of glyphosate-degrading Pseudomonas spp. strains isolated from contaminated soil. J Gen Appl Microbiol. 2015;61(5):165–70.
Andriani LT, Aini LQ, Hadiastono T. Glyphosate biodegradation by plant growth promoting bacteria and their effect to paddy germination in glyphosate contaminated soil. J Degrade Min Land Manage. 2017;5(1):995.
Wang GH, Berdy BM, Velasquez O, Jovanovic N, Alkhalifa S, Minbiole KP, Brucker RM. Changes in microbiome confer multigenerational host resistance after sub-toxic pesticide exposure. Cell Host Microbe. 2020;27(2):213–24.
Rubio F, Guo E, Kamp L. Survey of glyphosate residues in honey, corn and soy products. J Environ Anal Toxicol. 2014;5(249):2161–525.
Medici SK, Maggi MD, Galetto L, del Rosario IM, Sarlo EG, Recavarren MI, Salar PM, Eguaras MJ. Influence of the agricultural landscape surrounding Apis mellifera colonies on the presence of pesticides in honey. Apidologie. 2022;53(2):1–14.
Motta EV, Mak M, De Jong TK, Powell JE, O’Donnell A, Suhr KJ, Riddington IM, Moran NA. Oral or topical exposure to glyphosate in herbicide formulation impacts the gut microbiota and survival rates of honey bees. Appl Environ Microbiol. 2020;86(18):e01150-e1220.
Acknowledgements
'Not applicable'
Funding
The research was partially funded by the EU project “Nourishing PROBiotics to bees to Mitigate Stressors” (NO PROBleMS), H2020-MSCA-RISE 2017, GA 777760, 2018–2022 and by Fondo para la Investigación Científica y Tecnológica (PICT 2823–2017).
Author information
Authors and Affiliations
Contributions
GFDL, CB and PR collected bee samples. GFDL and SQ extracted the DNA from insect samples. GFDL, DA, LP and SQ analyzed the pathogen load in qPCR. GFDL, LP and DA performed the sequencing. DA and LB carried out bioinformatics on the microbiome raw data. MFDL and PR performed spatial analysis of land use. GFDL, MFDL, FZ and LB were involved in statistical analysis. ME and MM were involved in the research design. GFDL, DA, LP, ME, MM and DDG wrote the manuscript. ME, MM and DDG were involved in founding acquisition. All authors read and approved the final manuscript."
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical review and approval were waived for this study because the Italian law does not require and ethical approval for tests performed on arthropods with exceptions of cephalopods according to the Italian D.L. 4 March 2014 n. 26, and Italian implementing decree following the European regulation 2010/63/UE. Moreover, experimental honey bee colonies were purchased from a commercial beekeeper in Mar del Plata, Buenos Aires Province, Argentina. They were kept, under the regulation of the law Nº10.081/83. Art. 4, in the Social Bee Research Centre (CIAS) apiary, Universidad Nacional de Mar del Plata, Buenos Aires, Argentina. Although Ethical Committee authorization is not required for research with invertebrate animals, the researchers minimized the number of euthanized individuals as suggested by the UNMdP Bioethical Committee.
Consent for publication
'Not applicable'.
Competing interests
Authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Supplementary materials.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Fernandez De Landa, G., Alberoni, D., Baffoni, L. et al. The gut microbiome of solitary bees is mainly affected by pathogen assemblage and partially by land use. Environmental Microbiome 18, 38 (2023). https://doi.org/10.1186/s40793-023-00494-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40793-023-00494-w