
Abstract
Orthopedic conditions have emerged as global health concerns, impacting approximately 1.7 billion individuals worldwide. However, the limited understanding of the underlying pathological processes at the cellular and molecular level has hindered the development of comprehensive treatment options for these disorders. The advent of single-cell RNA sequencing (scRNA-seq) technology has revolutionized biomedical research by enabling detailed examination of cellular and molecular diversity. Nevertheless, investigating mechanisms at the single-cell level in highly mineralized skeletal tissue poses technical challenges. In this comprehensive review, we present a streamlined approach to obtaining high-quality single cells from skeletal tissue and provide an overview of existing scRNA-seq technologies employed in skeletal studies along with practical bioinformatic analysis pipelines. By utilizing these methodologies, crucial insights into the developmental dynamics, maintenance of homeostasis, and pathological processes involved in spine, joint, bone, muscle, and tendon disorders have been uncovered. Specifically focusing on the joint diseases of degenerative disc disease, osteoarthritis, and rheumatoid arthritis using scRNA-seq has provided novel insights and a more nuanced comprehension. These findings have paved the way for discovering novel therapeutic targets that offer potential benefits to patients suffering from diverse skeletal disorders.
Similar content being viewed by others
Background
Skeletal disorders are a major contributor to disability-adjusted life years, affecting 1.7 billion individuals worldwide who experience degeneration, fractures, and other orthopedic conditions [1,2,3,4,5,6]. These disorders can be attributed to aging, trauma, and immune factors, impacting various skeletal components such as bones (osteoporosis, osteopenia, etc.), joints [osteoarthritis (OA), rheumatoid arthritis (RA), etc.], spines (disc degenerative disease, ankylosing spondylitis, etc.), and muscles (sarcopenia, etc.) [7]. Not only do these conditions impair the patients’ work capacity and quality of life but they also impose a significant burden on the global medical system [8, 9]. In particular, prolonged and intense training regimens can significantly contribute to the development of skeletal disorders among military personnel, potentially leading to non-combat troop reduction [10]. Therefore, there is an urgent need for a comprehensive investigation of the pathogenesis and treatment approaches for these diseases. High-resolution research strategies are required to precisely elucidate the underlying mechanisms and develop more effective therapies for skeletal diseases [11].
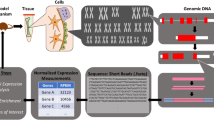
Over the past decade, single-cell RNA-sequencing (scRNA-seq) technology has emerged as a powerful tool for accurately examining the transcriptome at the resolution of individual cells [11,12,13,14,15,16] (Fig. 1). In contrast to bulk RNA sequencing, which provides an average measurement of gene expression across millions of cells, scRNA-seq generates sequencing libraries that map the transcriptome to individual cells, thereby clarifying the biological differences between cells [17, 18]. With its high throughput capabilities, scRNA-seq enables extensive gene profiling of more than 106 single cells per run, offering the potential to identify novel cell types and characterize molecular events within cellular subpopulations [19]. In recent years, scRNA-seq has been extensively applied to study skeletal disorders, shedding light on previously unexplored aspects of the skeletal micro-world [11, 20] (Fig. 1). These approaches have allowed researchers to examine the skeletal systems at an unprecedented resolution, enhancing our understanding of cellular heterogeneity and critical cellular events that govern skeletal homeostasis and disease [21,22,23,24]. Furthermore, scRNA-seq has facilitated the elucidation of the intricate molecular network involved in intercellular crosstalk, providing crucial insights into the cellular microenvironment that often contributes to pathological processes [25,26,27,28,29].

Single-cell RNA sequencing (scRNA-seq) reveals the cellular heterogeneity in unprecedented resolution in skeletal research. a scRNA-seq can dissect the cellular composition of specific skeletal tissues in different conditions, providing strategies of prospective isolation for target cell populations using fluorescence-activated cell sorting (FACS). b Differential expression analysis of scRNA-seq data helps identify both classic and novel characteristics of cell clusters. c The fate of cells can be predicted using single-cell trajectory inference methods, which map the developmental pathways of cells based on their gene expression profiles. d The relationships and intercellular communications among different cell clusters can be predicted through scRNA-seq data, which is crucial for understanding tissue function and disease progression
Despite the numerous advantages of scRNA-seq, several challenges persist in various aspects. Firstly, it is difficult to obtain high-quality single cells from bone and cartilage tissues with dense collagen and high mineralization, often failing to meet the criteria for scRNA-seq [30]. Secondly, the diverse cell morphologies within the skeletal system necessitates careful selection of an appropriate sequencing method [30]. Lastly, inadequate bioinformatic analyses without stringent quality control measures may undermine data mining efforts. This review summarizes significant advancements in skeletal research, including sampling processing, sequencing methods, and basic bioinformatic analysis, while also presenting illustrative examples that demonstrate the full potential of scRNA-seq in skeletal studies. Additionally, the integration of scRNA-seq with multi-omic strategies may provide benefits for bridging the knowledge gaps.
Single-cell acquisition approaches in skeletal tissues
The emergence of single-cell sequencing technology has revolutionized the field of skeletal research, providing a powerful tool to explore cellular diversity and unravel the intricate molecular mechanisms underlying complex diseases.
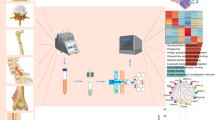
The success of single-cell sequencing technology heavily relies on high-quality techniques for capturing individual cells, which have gained significant traction in recent years [11, 31]. Numerous single-cell acquisition technologies are used to acquire high-quality single-cell suspensions from skeletal tissues, which can be divided into mechanical dissociation, enzymatic digestion, and cell population enrichment including fluorescence-activated cell sorting (FACS), magnetic bead sorting (MACS), and microfluidics [32,33,34] (Fig. 2). To obtain a high-quality single-cell suspension, we recommend following a streamlined approach as outlined below: (1) Mechanical dissociation. This step involves breaking down the tissue to separate the cells and enhance their accessibility for enzymatic digestion, facilitating the release of individual cells from the tissue matrix. (2) Enzymatic digestion. Following mechanical dissociation, enzymatic digestion is performed to degrade the extracellular matrix. This step is essential for separating cells from the tissue scaffold and ensuring the production of a single-cell suspension. (3) Cell population enrichment. This step employs techniques to isolate specific target cells from the heterogeneous cell suspension. Methods such as FACS or MACS can be utilized to selectively isolate desired cell populations based on molecular markers or other criteria.
Mechanical dissociation
Mechanical dissociation is a vital technique for single-cell isolation in skeletal studies [35] (Fig. 2a). It involves microdissection to separate tissue structures, mincing using tools like razor blades and surgical scissors tailored to specific tissue types, and grinding with digestion buffer [36]. This method is crucial for preparing samples for single-cell sequencing [37], as seen in studies on bone marrow cells where it highlights a dynamic and heterogeneous molecular landscape that exhibits high responsiveness to stress [38]. Maintaining low temperatures throughout the process is essential to preserve cell integrity and protein functionality.

Overview of single-cell acquisition technologies used to acquire high-quality single-cell suspensions from skeletal tissues. a Mechanical dissociation involves techniques such as microdissection, sectioning, and grinding to physically separate cells. b Enzymatic digestion is an effective method that utilizes enzymes like trypsin and collagenase to break down the extracellular matrix, facilitating cell separation. DNase is also employed to extract free DNA from cell clusters. c (i) Fluorescence-activated cell sorting is a pivotal and precise technology for isolating single cells through fluorescence markers. (ii) Magnetic bead sorting employs magnetic beads to acquire single-cell. (iii) Microfluidic technologies represent advanced methods for single-cell acquisition. These technologies are notable for their compact design, high throughput, and enhanced sensitivity, making them indispensable in modern cellular biology
Enzymatic digestion
The enzymatic digestion method uses specific enzymes to degrade the extracellular matrix of cells, effectively separating cellular components. This technique is highly efficient for single-cell separation, cost-effective, easy to operate, and minimally impairs cell integrity (Fig. 2b). For skeletal tissue with a complex extracellular matrix, the combination of collagenase and DNase I can enhance the viability of bone cell component and improve the efficiency of the separation process [36]. Collagenase digestion is the primary method applied for chondrocyte isolation by breaking down the cartilaginous extracellular matrix (ECM). Typically, three rounds of digestion lasting approximately 8 h are required to fully digest the cartilage ECM and obtain high-quality chondrocytes [39]. In the case of intervertebral discs (IVDs), a recommended combination of trypsin, protease, and collagenase is used for about 3.5 h (0.5 h for trypsin, 1 h for protease, 2 h for collagenase) to effectively break down collagen fibers and ECM to yield single cells [23]. Enzyme digestion is commonly employed as a method for isolating individual muscle or tendon cells. By incubating muscle tissue with a combination of collagenase type II, collagenase D, and dispase II for 1 h, myocytes can be effectively dissociated from other components [40].
Although the enzymatic digestion method holds promising prospects for single-cell sequencing in the skeletal system, careful consideration should be given to tissue type, enzyme combination, and enzyme concentration to determine the appropriate digestion time and ensure reproducibility and reliability. By utilizing the enzymatic digestion method, bone cell components can be obtained quickly and efficiently for subsequent analysis using scRNA-seq.
Cell population enrichment
FACS is a critical technology for single-cell isolation, enabling precise cell sorting through fluorescence markers (Fig. 2c). It’s widely used in fields such as oncology and skeletal research, where it aids in single-cell sequencing [41,42,43]. For instance, Mo et al. [25] employed FACS to isolate specific skeletal cells for scRNA-seq studies on cell maintenance and lineage. However, the use of FACS may induce cellular stress that can affect sequencing accuracy, and requires specialized equipment and expertise, making it expensive and technically demanding [44]. Its resolution limitations also pose challenges in distinguishing similar cell types, with results potentially influenced by environmental and operational factors.
MACS is another widely utilized technique for single-cell acquisition, employing magnetic beads that bind to cell surface antigens or specific intracellular substances for separation in a magnetic field (Fig. 2c). This method is highly regarded for its rapidity, efficiency, and minimal cellular damage, making it the preferred choice for single-cell sequencing in skeletal research. For instance, studies have used MACS to isolate stromal cells from murine bone marrow to explore their roles in the hematopoietic stem cell niche and identify distinct subpopulations of bone marrow-derived mononuclear cells for lineage-specific investigations [45, 46]. Despite offering numerous advantages, meticulous sample preparation is necessary with MACS to prevent cellular damage and ensure precise cell sorting, and the bead binding has the potential to alter the biological properties of the cells.
Microfluidic technology is increasingly utilized in single-cell sequencing due to its compact size, high throughput, and sensitivity (Fig. 2c). It excels in the isolation, sorting, and sequencing of individual cells, facilitating efficient capture and detailed transcriptome analysis. The C1 Single-Cell Auto Prep System has been effectively employed for SMART-Seq of dendritic cells, highlighting its capability to detect transcriptomic variations and rare cellular responses [47]. Although microfluidic technology has several advantages over other single-cell acquisition methods, a critical limitation of Fluidigm microfluidic chips is their size restriction on captured cells (the largest chip is designed for cells up to 30 μm) that can undergo single-cell analysis [48].
The field of skeletal systems has been revolutionized by the advent of single-cell acquisition technology, which enables the investigation of cellular heterogeneity and intricate molecular mechanisms. Through high-quality single-cell capture technology, researchers can isolate individual cells from various sources such as bone marrow, cartilage, bones and joints. However, it is important for researchers to carefully consider both the advantages and limitations of these techniques such as reliance on cell characteristics and potential cell damage to determine the most appropriate approach for single-cell acquisition in their specific applications.
scRNA-seq platforms applicable to the skeletal system
It is important to choose the appropriate library construction strategy for target tissues [37, 49]. Large-scale scRNA-seq methods such as the drop-seq-based 10× Genomics Chromium system, microwell-seq-based BD-Rhapsody system, and the DNBelab C4 system from MGI, can efficiently isolate numerous cells from hypercellular tissues [50,51,52]. For tissue with low-input cells, Smart-seq2 and CEL-seq2 are more applicable for capturing the single-cell transcriptome at a high sequencing depth using automated micropipettes or FACS [53]. In this section, we present the advantages of different strategies in terms of throughput, sequencing depth, and application scope (Table 1).
10× Genomics chromium system
The 10× Genomics Chromium system is one of the most widely used sequencing platforms. Specifically, cells are encapsulated in microfluidic devices and labeled using barcoding technology, leading to a significant reduction in time and cost [50, 54]. The highly parallelized nature of this system allows for the sequencing of up to 80,000 cells per sample. Moreover, its exceptional level of automation enables it to handle all aspects of the cellular suspension preparation process, single-cell capturing, library amplification, and fragment tagging within a matter of hours. Therefore, the 10× Genomics Chromium system has gained considerable popularity in skeletal research. For example, Huang et al. [55] applied this method to decode a staggering number of 102,077 cells from knee joint tissues and successfully illustrated the cellular heterogeneity associated with OA. Liu et al. [56] performed scRNA-seq on 100,987 osteosarcoma cells and revealed valuable insights into immune cell functions in recurrent and metastatic pulmonary osteosarcoma lesions. However, probabilistic capture may result in potential loss or underrepresentation of rare cell clusters, which should be taken into account.
BD-Rhapsody system
Similar to the 10× Genomics Chromium system, the BD-Rhapsody system is also capable of efficiently handling large numbers of cells. The cell capture process in the BD-Rhapsody system is carried out using microwell precipitation, known as CytoSeq, which has over 200,000 micropores in a single plate [51]. The CytoSeq improves the probability of successful cell capture and improves the viability tolerance of cells within the BD-Rhapsody system. Moreover, the cDNAs bound to the beads can be stored at 4℃ for up to 3 months, enabling users to pool libraries from multiple samples for sequencing. For example, Tu et al. [27] applied the BD-Rhapsody system to decode human IVDs at different degenerative levels. Zhang et al. [57] utilized this technology to capture cells in bone fractures and elucidated the role of B cells in fracture repair processes. However, due to the limitation of bead binding efficiency, capturing cells with diameters greater than 20 μm may experience a significant decrease in efficiency when using the BD-Rhapsody system.
Smart-seq2 and CEL-seq2
Smart-seq2 and CEL-seq2 are low-throughput strategies that use automated micropipettes or FACS to isolate cells into 96-well or 384-well plates [53]. These methods improve the sequencing depth of individual cells and are typically used for dissecting heterogeneity in specific cell populations [53]. Smart-seq2 captures the whole transcriptome, while CEL-seq2 only retains the sequences from the 3’ end of mRNAs, thus it is also subject to limitations due to 3’ bias [58, 59]. In addition, CEL-Seq does not detect miRNAs and other nonpolyadenylated transcripts. While this can be regarded as an advantage for enhancing mRNA level measurement efficiency, it also poses a disadvantage by failing to detect rRNA [60]. Hedlund et al. [61] employed Smart-seq2 to investigate the dynamic changes of neural stem cells after spinal cord injury based on a population of 487 GFP+ Nestin+ cells. Mizoguchi et al. [62] utilized Smart-seq2 to analyze the synovium of OA patients and identified an invasive fibroblast population located in the perivascular area of inflamed synovium. Zhang et al. [26] used CEL-seq2 to examine immune cells in the synovium of OA. In contrast to Drop-seq strategies, Smart-seq2 or CEL-seq2 can detect more genes in an individual cell. Smart-seq2, in particular, exhibits the superior ability to detect gene expression, which is particularly advantageous for cell types with low abundance transcripts such as terminally differentiated chondrocytes [63].
Given the diversity present in the skeletal tissues, it is advisable to first optimize cell isolation procedures and disentangle cellular complexity before subsequent analysis. This is especially important when conducting large-scale transcriptomic profiling and exploring rare but significant cells during bone development and related disease progression.
Practical scRNA-seq analysis pipelines of exploring cellular heterogeneity in skeletal tissues
The raw reads obtained from Next Generation Sequencing machines undergo pre-processing, which includes data cleaning, adapter trimming, and genome mapping. These steps can be performed individually or integrated into software such as Cellranger developed by 10× Genomics. The final output is a gene matrix that is subjected to well-established analysis workflows, including Seurat (implemented in R) [64] and Scanpy (implemented in Python) [65]. While these two powerful analytic tools can manage most scRNA-seq datasets, customized analyses are required for specific projects involving various combination strategies such as differential trajectory simulation and intercellular communication networks [66, 67]. Typically, scRNA-seq enables the simultaneous revelation of transcriptomic features across all cell populations and the comprehensive prediction of their potential functions in disease progression. More importantly, targeting signature genes and enriched pathways in key cell types provides potential therapeutic targets for clinical applications. Here, we introduce the practical bioinformatic analysis steps for scRNA-seq in skeletal research.
Quality control (QC)
QC serves as the initial step in the scRNA-seq analysis pipeline, aiming not only to eliminate low-quality data that may interfere with downstream analysis but also to exclude non-biological factors introduced by experimental conditions such as RNA degradation, elevated mitochondrial gene rates, and digestive stress. Additionally, QC can reduce noises induced by the sequencing system, such as doublets and multiplets. A rigorous QC process is crucial for filtering out low-quality cells. However, thresholds for QC should be set carefully when integrating datasets due to variations in gene expression levels among different cell types.
The basic criteria for QC involve gene numbers, count numbers, and mitochondrial gene rates. Various packages implemented in R (e.g., scuttle, DoubletFinder, DoubletDecon) and Python (e.g., scrublet, and DoubletDetection) are employed to remove potential doublets and multiplets [68,69,70,71]. After completing the QC process successfully, matrices should be normalized to correct relative gene expression abundances and facilitate cell comparison for further analysis.
Batch effect removal
The use of different timepoint, equipment, reagents for cell capture, and even operating personnel can inevitably lead to batch effects among datasets. More than 50 integration strategies have been reported for benchmarking scRNA-seq datasets [72, 73]. Among them, mutual nearest neighbors (MNN) or FastMNN, Seurat v3 integration, Harmony, MNN, and scGen are the most commonly used methods [74,75,76,77]. In our previous scRNA-seq datasets of human IVD cells, we compared the performances of these methods [23], and found that FastMNN and scGen showed a better balance between removing batch effect and retaining dimensional structure (Fig. 3). Huang et al. [55] applied Seurat v3 to integrate scRNA-seq datasets derived from 5 OA patients and uncovered 7 distinct populations. With the rapid increase in single-cell sequencing data on skeletal maintenance and disorders from different laboratories and platforms, effective data integration plays a crucial role in analyzing cellular heterogeneity and identifying key clusters responsible for the disease. However, it is important to avoid overcorrection during batch effect removal to preserve the biological signatures. Multiple integration methods may need to be evaluated to reveal the major features embedded in the datasets.

Performance of different batch effect removal strategies in the integration of scRNA-seq datasets on human IVD cells. a Evaluation, applicable programming language and website of FastMNN, Seurat v3 (CCA), Harmony and scGen methods. b The dimensionality reduction plots of raw data, FastMNN, Seurat v3 (CCA), Harmony and scGen contain two rows. In the first row, cells are colored by different sites of intervertebral disc, and in the second by cell type. Seurat v3, Harmony are embedded in t-SNE, and FastMNN, scGen are embedded in UMAP. Each method can well eliminate the batch effect while FastMNN and scGen have better performance [23]. Copyright © 2021, Published by Springer Nature. AF annulus fibrosus, Chond chondrocyte, CEP cartilaginous endplate, FastMNN fast mutual nearest neighbors, NPPC nucleus pulposus progenitor cells, t-SNE t-distributed stochastic neighbor embedding, UMAP uniform manifold approximation and projection, PCA principal component analysis, CCA canonical correlation analysis, NP nucleus pulposus, IVD intervertebral disc, Noto notochord cell, EC endothelial cell
Dimensionality reduction
The first step in reducing the dimensionality of high-dimensional datasets is to identify the principal components by calculating the highly variable features. To create informative and visually appealing graphs, several well-developed algorithms are used for dimensionality reduction (DR) and visualization, including principal component analysis (PCA), t-distributed stochastic neighbor embedding (t-SNE) [78], and uniform manifold approximation and projection (UMAP) [79].
PCA captures the data variance through a linear combination of gene expression levels. Its simplicity and efficiency make it usually the initial round of DR in scRNA-seq analysis, although explaining the complete polynomial relationship among features in large and complex datasets may be challenging. t-SNE and UMAP are widely used nonlinear dimensionality reduction algorithms that project the high-dimensional structural features to low-dimensional space. UMAP displays a more realistic global distribution because of its loss function and distance algorithm, whereas t-SNE retains complete local distribution information. Moreover, the UMAP algorithm has lower time consumption than t-SNE, making it faster for scaling large datasets. However, a recently developed algorithm called FFT-accelerated interpolation -based t-SNE can accelerate calculation by over 10-fold if high memory consumption on the machine is tolerable. Finally, t-SNE uses random distribution to initialize low-dimensional data, while UMAP employs the Laplace transform to assign the initial low-dimensional coordinates, resulting in higher stability of UMAP results. In orthopedic research, many studies prefer using the t-SNE reduction method to obtain a fuller composition [80,81,82,83,84].
Clustering and annotation
Clustering is a crucial step in customized analysis, enabling the exploration of cellular heterogeneity and the identification of novel populations. Among various clustering methods, K-means is widely adopted due to its ability to assign each cell to the nearest center point [85]. However, being a greedy algorithm, it cannot guarantee finding the overall optimal solution, especially for clusters with rare cells [15]. In Seurat and Scanpy workflows, Louvain is a default clustering algorithm based on modularity or network graphs [86, 87]. Nonetheless, it has limitations in some situations where cluster portraits are influenced by variations in cellular distribution density within clusters [88,89,90]. To address these issues, Traag et al. [90] developed an optimized Leiden algorithm that offers faster running speed and ensures community connection, thereby rationalizing the process of clustering.
The strategy for defining a cluster should encompass comprehensive recognition. Typically, differentially expressed genes (DEGs) among clusters serve as signatures for each cluster. Based on this, previous studies have successfully identified various cell types in skeletal tissues [23]. Conveniently, population definition can be performed by automatic annotation packages, such as SingleR [91], Cellassign [92], Garnett [93], and scTPA [94]. It is recommended to adopt a combined strategy that considers identified marker genes and postulated cell types from annotation packages.
In many cases, the identification of a cell cluster cannot be determined by a limited number of the signature genes. Therefore, biological processes or pathways enriched by the DEGs can assist in defining cell identity. Enrichment analyses including Gene Ontology [95], Kyoto Encyclopedia of Genes and Genomes [96], and Gene Set Enrichment Analysis (GSEA) [97] are commonly employed for cluster annotation purposes. For instance, chondrocytes and fibroblasts are always associated with extracellular matrix organization, while chondrocytes exhibit closely related to the transforming growth factor-β (TGF-β) signaling pathway, and osteoblasts are linked to ossification and the secreted phosphoprotein 1 (SPP1) signaling pathway [20, 98].
Usually, known cell markers in top DEGs for each cell type are used for annotating cell clusters. Nevertheless, there may be novel genes that show significant expression levels, indicating their potential as new molecular markers for the cell cluster. These potential markers can be further validated through rigorous experiments such as in situ hybridization or immunofluorescence staining to confirm their efficacy in identifying cell types. Previous research has demonstrated that these newly identified markers can help identify key cell clusters and facilitate prospective isolation of specific cell clusters involved in skeletal development, maintenance, and disease progression [23, 99].
Regulatory network
Gene regulatory networks (GRN) are responsible for determining and maintaining the transcriptional state of cells, making them an essential step in understanding cell states. The computational method single-cell regulatory network inference and clustering (SCENIC) is used to reconstruct GRN and evaluate their activity in each cell for identifying cell states [100]. Compared with other co-expression algorithms, SCENIC utilizes RcisTarget to identify potential direct-binding targets and exclude false positive outcomes [100]. Despite its effectiveness in analyzing specific and robust GRN underlying cell states, there are certain limitations associated with SCENIC, such as increased time-consuming when dealing with large datasets. It is advisable to infer the GRN from a subsampled dataset and include all cells during the scoring step, or opt for a more efficient algorithm such as GRNBoost [100]. Studies using SCENIC have revealed the regulators involved in enthesis progenitor cell differentiation and mineralization including SOX9 and RUNX2 [101], as well as common regulatory programs within the pathogenic subset of both articular and meniscus tissues that encompass key members of the CREB family and ZEB1 [102]. Additionally, SCENIC has been used to decipher the programs governing chondrocytes and the pericyte-like cell cluster [103]. In our previous study, SCENIC was employed to uncover the GRN that determined cell fate in the nucleus pulposus progenitor cells (NPPC) subclusters, revealing strong enrichment of SMAD3 in the PROCR+ NPPC-3 cluster [23]. Overall, the usage of SCENIC will provide invaluable biological insights into the mechanisms driving cellular heterogeneity.
Differentiation trajectory inference
Pseudotime analysis is used to illustrate the lineage development or differentiation processes of specific cell types. By identifying the dynamic changes of gene expression along the trajectory, the cell state space metric can be simulated in silico. Two major tools widely used in pseudotime analysis are Monocle and RNA velocity. Monocle, proposed by Cole Trapnell et al. [104] reconstructs linear trajectories using a minimum spanning tree (MST) algorithm. RNA velocity, proposed by La Manno et al. [105], calculates the derivative of unspliced RNA to spliced RNA to obtain the kinetics of mRNA cycles and infer differentiation vectors of single cells. Commonly used RNA velocity tools include velocyto [105] and scvelo [106].
It is important to note that the MST algorithm has an advantage in trajectory construction but depends on prior knowledge when specifying the root of differentiation. In contrast, RNA velocity can predict the direction of the cell lineage based on real transcript dynamics without the knowledge of the development process. However, the result of RNA velocity largely depends on the previous dimensionality reduction diagram. He et al. [22] utilized RNA velocity to simulate chondrogenesis and osteogenesis in human embryonic skeletal development and demonstrated a subset of skeletal stem and progenitor cells that play crucial roles in the fate decision of limb bud mesenchymal differentiation. Our previous study applied Monocle3 to decipher the chondro-osteogenic potential of NPPC in human IVDs [23].
Intercellular communications interface
The maintenance of skeletal tissues largely depends on the homeostasis of the microenvironment, which is regulated by complex intercellular communications. Therefore, analyzing these communications is critical for uncovering the molecular mechanism underlying disease. By examining the co-expression of ligand-receptor genes, it becomes possible to predict key pathways involved in regulating cell functions and identify potential targets for intervention. CellPhoneDB [107] and CellChat [108] are widely used tools in skeletal research. Wu et al. [109] applied CellPhoneDB to demonstrate that clusters of dendritic cells, T cells, and macrophages observed in RA patients display enhanced interactions mediated by molecules such as CD74, and CCL13, which may contribute to the abnormal inflammatory responses seen in RA. Ling et al. [110], using CellChat, revealed that M2 macrophages can modulate ProNPC function through macrophage migration inhibitory factor (MIF) and TGF-β signaling.
Various applications of scRNA-seq research in skeletal health and disease
Crucial cell clusters and molecules guiding the development and degeneration in the spine
The process of spinal development initiates with the differentiation of the sclerotome from the somite. The first pair of somites emerges around day 20 of embryonic development, followed by subsequent pairs forming at a rate of three pairs per day, which is regulated by a molecular oscillator known as the segmentation clock [111, 112]. To confirm the presence of a similar oscillator in humans, Diaz-Cuadros et al. [113] performed scRNA-seq on mouse and human pre-somatic mesoderm cells, revealing a comparable developmental trajectory and supporting the existence of a human segmentation clock. However, there is still limited understanding regarding gene expression dynamics during spinal development. Li et al. [114] conducted scRNA-seq on a pig embryo at 27-day post-fertilization, equivalent to a human embryo at approximately 9 weeks gestation. By using monocle and RNA velocity analysis methods, they constructed 2 distinct trajectories for angiogenesis and osteogenesis, while identifying 6 cell clusters. Notably, HOXA10 expression was predominantly restricted to lumbar vertebrae clusters, suggesting its role as a determining factor in lumbar formation. Yu et al. [115] analyzed the transcriptome of human fetal spines from 8 to 17 gestational weeks at single-cell resolution and found that HIST1H1A+COL2A1− fibroblasts may be regulated by TUBB along with its upstream transcription factor HOXA10. Recently, an integrated analysis combining spatiotemporal information and scRNA-seq was performed on both human and mouse spines during embryonic stages. This study also incorporated lineage tracing techniques to identify 2 types of notochord-derived nucleus pulposus (NP) cells responsible for IVD formation [116]. Collectively, the above studies provide comprehensive insights into cellular heterogeneity and regulatory processes involved in spine formation as revealed by scRNA-seq.
Following birth, the spines bear the weight of the body and the loads associated with movement. IVDs are crucial for maintaining the mechanical properties of the spine due to their elasticity and resilience. Single-cell RNA sequencing unveiled distinct cell types including progenitor cells and chondrocytes in healthy IVDs and critical biological processes including matrix changes and immune activation during degenerative conditions (Fig. 4a). Gao et al. [99] performed scRNA-seq on postnatal mouse IVDs and found UTS2R+ nucleus pulposus progenitors (ProNPs) that have trilineage differentiation potential ex vivo. They also discovered the specific expression of tenascin-C (TNC) in ProNPs and confirmed its role in promoting adhesion and inhibiting apoptosis by ex vivo studies [99]. In our previous study using scRNA-seq, we revealed various cell clusters in healthy IVDs, including chondrocytes, notochord cells, endothelial cells, and pericytes [23]. These chondrocytes were further categorized into regulatory, homeostatic, and effector subpopulations with distinct ECM properties. We also identified PROCR+PDGFRA+ NPPCs with potential stemness. Monocle3 analysis demonstrated two differentiation fates towards osteogenesis and chondrogenesis, which were subsequently confirmed ex vivo. Additionally, we used CellChat analysis to establish an intercellular communication network and identified the key regulatory molecules TGF-β and PDGFRA, which regulate NPPC chondrogenesis and proliferation. Apart from NP cells, Wang et al. [117] found Lepr+ annulus fibrosus (AF) stem cells in the intervertebral stem cell niche, expressing stemness markers like CD105. These cells differentiated into fibro-chondrocyte-like AF cells in vitro (Fig. 4a). These scRNA-seq studies have provided evidence for the presence of IVD progenitor cells along with their significant functions in maintaining homeostasis.
Spine disorders lead to severe back pain and mechanical dysfunction, significantly impairing the quality of human life. Degenerative disc disease (DDD) is a prevalent cause of low back pain [2], which is associated with microenvironmental disorders and alterations in cellular heterogeneity. Han et al. [118] conducted scRNA-seq on normal, mildly degenerative, and severely degenerative NPs, revealing an increasing inflammatory response in cartilage progenitor cells following degeneration. Similarly, Ling et al. [110] observed an inflammatory response and an increase in fibrocartilaginous NP cells, while metabolic and homeostatic NP cells decreased after degeneration in humans. Besides the enhanced inflammatory response of chondrocytes, scRNA-seq also showed an increase in EGNL3+ StressCs, but a decrease in TGFBR3+ HomCs and GPRC5A+ RegCs in degenerative goat IVDs, which was further validated in vitro [119]. Li et al. [120] identified inhibitory calcified chondrocytes, fibrochondrocytes, and calcified chondrocytes with high expression levels of MGP, COL1A1, and FN1 that were more abundant within degenerative NP as well. Zhang et al. [121] found upregulated genes that related to the ferroptosis pathway in chondrocytes after degeneration. The rigid extracellular matrix represents another critical pathological feature of disc degeneration. Zhou et al. [122] revealed that matrix stiffness could activate the YAP/TEAD1-Cyclin B1 axis to promote proliferation of NP cells and IVD fibrosis, and scRNA-seq shed light on YAP+ Fibro NPCs as the key subcluster involved in IVD pathological fibrosis (Fig. 4b). These insights into alterations occurring in degenerative chondrocytes provided by scRNA-seq contribute to a better understanding of the mechanisms underlying function degradation seen in DDD.

Single-cell RNA sequencing unveiled distinct cell types including progenitor cells and chondrocytes in healthy intervertebral discs (IVDs) and critical biological processes including matrix changes and immune activation during degenerative conditions. a In healthy IVDs, a variety of cells including progenitor cells, chondrocytes, and notochord cells play crucial roles in maintaining the IVD homeostasis. b In degenerated IVDs, there is a notable alteration in the phenotypes of progenitor cells and chondrocytes. The stiffening of the matrix activates YAP/TAZ signaling pathways, which in turn promotes chondrocyte proliferation and contributes to IVD fibrosis. Concurrently, macrophages and T cells not only proliferate but also engage in active crosstalk, influencing inflammatory responses. Additionally, the number of G-MDSCs increases, which plays a role in inhibiting matrix degeneration and suppressing T cell proliferation. AF annulus fibrosus, AFSC annulus fibrosus stem cell, Chond chondrocytes, NPC nucleus pulposus cell, NPPC nucleus pulposus progenitor cells, ProNP nucleus pulposus progenitors, TNC tenascin-C, TGF-β transforming growth factor-β, PDGFRA platelet-derived growth factor receptor alpha, FTL ferritin light chain, Fibro fibrogenic, MMP13 matrix metallopeptidase 13, YAP Yes-associated protein, TAZ tafazzin, TEAD1 TEA domain transcription factor 1, CCNB1 G2/mitotic-specific cyclin-B1, CTGF connective tissue growth factor, G-MDSCs granulocyte-like myeloid derived suppressor cells, UTS2R urotensin-2 receptor, PROCR protein C receptor, LEPR leptin receptor, SFMA/HAMA/FCI methacrylated SF/methacrylated HA/fibrochondrogenic inductive, HO-1 heme oxygenase-1
As degeneration progresses, microvasculature forms when immune cells are activated and infiltrate the IVDs [123]. This phenomenon has been validated by numerous studies [27, 110, 118, 119, 124,125,126,127,128]. Tu et al. [27] identified granulocytic myeloid-derived suppressor cells in degenerative NP and confirmed their role in inhibiting T cells and alleviating matrix degradation in vitro. The interactions between immune cells and IVD cells at single-cell resolution have been revealed through CellPhoneDB analysis, and the successful reversal of NP ossification was achieved by inhibiting TNF-a. This was verified by Guo et al. [128] using a rat coccyx disc degeneration model. In addition, the exhaustion of stem/progenitor cells contributes to DDD. Gao et al. [99] identified UTS2R+ ProNPs that were found to be exhausted in degenerative IVDs; however, their transplantation with TNC can attenuate the progression of DDD. Wang et al. [117] established an SFMA/HAMA/FCI composite hydrogel laden with AFSC, which showed a strong repair capacity for injured AF. PROCR+ PC was conserved in goat IVDs and exhibited a differentiation program alteration to stemness exhaustion [119] (Fig. 4b). scRNA-seq revealed the role of immune cells in stem/progenitor cell exhaustion in DDD, providing valuable insights for potential therapeutic interventions.
Besides DDD, ankylosing spondylitis (AS) is another prevalent spine disease. Approximately 80% of patients with AS experience symptoms before the age of 30 years [129]. Although the etiology remains unknown, AS is widely considered a chronic inflammatory disease [129]. Xu et al. [130] compared the heterogeneity of peripheral blood mononuclear cells (PBMCs) from healthy individuals and AS patients, revealing an increase in naïve CD8+ T cells, CD8+ T cells, memory CD4+ T cells, and memory B cells in AS patients with elevated expression of genes associated with the inflammatory pathway. On the contrary, Ren et al. [131] observed a decrease in natural killer (NK) cells and a bias towards CD56bright NK cells in the PBMCs from AS patients. Cribbs et al. [83] performed scRNA-seq on demethylase inhibited Th17 cells, revealing a shift from highly inflammatory cell subsets toward a resting state due to the reduced expression of key metabolic transcription factors, thereby providing a potentially effective therapeutic target for AS and other autoimmune diseases. As an autoimmune disease itself, there are strong associations between AS and Crohn’s disease [132]. Lefferts et al. [133] found that the PBMCs from patients suffering from both conditions showed a significant increase in granzyme B+ T cells and greater T cell maturity levels were observed. In addition, the pathological mechanism of AS involves Fibroblasts. The study by Li et al. [134] demonstrated that TNC suppressed ECM adhesion force by activating the downstream Hippo pathway signaling, which subsequently increased chondrogenic gene expression, resulting in new bone formation at entheses sites. Using scRNA-seq techniques identified a cluster of FSP1+ fibroblasts secreting TNC, thereby facilitating our understanding of AS pathogenesis [134].
Despite its advantages in spinal research, scRNA-seq has limitations including the potential loss of rare cell populations due to the low cell density in IVDs, particularly the NPs. To address these challenges, further improvements in technology and larger tissue samples for library constructions are needed.
Cell dysfunctions contribute to the pathogenesis of OA and RA in joint
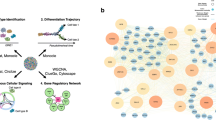
Joints facilitate the movement and flexibility of limbs by enabling synovium development, which initiates the formation of an intermediate zone. In this zone, the Gdf5-expressing lineage actively participates in joint tissue formation and establishes a cohort of progenitor cells with co-generation capabilities [135]. To elucidate the underlying mechanism, Bian et al. [24] conducted scRNA-seq on mouse embryo knee joints and classified three clusters of Gdf5-lineage enriched cells in different developmental states through RNA velocity analysis. Pseudotime and immunofluorescence analysis further revealed the transcriptional profiles of the major developmental paths for joint progenitors [24]. Combining scRNA-seq and lineage tracking, Collins et al. [136] also found that chondrocyte injury led to lining hyperplasia, due to the proliferation and differentiation of Prg4-expressing progenitors into fibroblast-like synoviocytes. Further differentiation trajectory analysis demonstrated that Sox5 and Foxo1 were the key transcription factors of fibroblast-like synoviocytes in mice and humans [136]. Besides, Gao et al. [137] performed scRNA-seq on murine hindlimbs at postnatal day 1, 5, 14, and 28 to systematically dissect the developmental process. They identified CD34 and Ly6e-positive candidate progenitors in articular cartilage and enthesis, as well as 3 cellular developmental branches marked by Col10a1, Spp1, and Tnni2 in the growth plate [137] (Fig. 5a). These results highlight the importance of progenitor cells in the joint formation and homeostasis.

Single-cell RNA sequencing analysis elucidated critical alterations during the progression of osteoarthritis. a Different clusters of progenitor cells in distinguish stages of mice joints. b In osteoarthritis, chondrocytes undergo senescence and ferroptosis processes that contribute to cell aging and death. This cellular deterioration is associated with increased pain sensitivity in the affected joints. c Fibroblasts, and immune cells are triggered in osteoarthritis and improve inflammation, neuronal growth and chondrocyte senescence. OA osteoarthritis, ZEB1 Zinc finger E-box binding homeobox 1, FAP fibroblast activation protein, ECM extracellular matrix, TRPV1 transient receptor potential cation channel subfamily V member 1, GPX4 glutathione peroxidase 4, GSSG glutathione disulfide, GSH glutathione, EAAT solute carrier eamily 1 member 3, PIEZO2 piezo-type mechanosensitive ion channel component 2, Ntrk 1 high affinity nerve growth factor receptor, GDF5 growth/differentiation factor 5, CDKN2A cyclin-dependent kinase inhibitor 2 A, SERPINE1 plasminogen activator inhibitor 1, CHI3L1 chitinase-3-like protein 1, CD cluster of differentiation, RARRES2 retinoic acid receptor responder protein 2, LGALS1 galectin-1, Ly6e lymphocyte antigen 6E, Prg4 proteoglycan 4, Rspo2 r-spondin-2, IL interleukin, IGF2BP3 insulin-like growth factor 2 mRNA binding protein 3
Over time and with accumulated joint motion load, articular degenerative diseases such as OA can develop. Single-cell RNA sequencing analysis elucidated critical alterations during the progression of osteoarthritis (Fig. 5). OA is a complex condition that affects the whole joint, involving chondrocytes, fibroblasts, and immune cells in its pathogenesis [138,139,140,141]. Ji et al. [28] identified 7 distinct populations of chondrocytes in the human OA cartilage. Through GSEA, they revealed 3 novel phenotypes, effector chondrocytes (ECs), regulatory chondrocytes, and homeostatic chondrocytes, which are primarily involved in metabolism, signaling pathways, and modulating cellular homeostasis respectively. Pseudotime trajectory analysis determined potential transition among proliferative chondrocytes, prehypertrophic chondrocytes, and hypertrophic chondrocytes [28]. The identification of these cell populations greatly enhances our understanding of the pathological mechanism underlying OA. Chou et al. [142] found that intact cartilage primarily consists of homeostatic and hypertrophic chondrocyte subpopulation, while damaged cartilage is enriched with prefibrotic- and fibrotic-, regulatory-, reparative- and prehypertrophic-chondrocytes. Subsequently, Wang et al. [143] observed an expansion of CHI3L1+ RegCs in OA, while Li et al. [144] demonstrated the regenerative capacity of CHI3L1+ chondrocytes. Hu et al. [145] uncovered that fibrous cartilage degeneration is primarily induced by fibrocartilage chondrocytes, and ECs were found to predominantly exert immune function in OA. Compared to intact cartilage, the intercellular communication between different chondrocyte subclusters within damaged cartilage was enhanced thorough pleiotrophin (PTN), nicotinamide phosphoribosyltransferase (NAMPT), SPP1, TGF-β and other signaling pathways as indicated by CellChat analysis [146] (Fig. 5b). A comprehensive analysis focusing on dysfunction in these specific chondrocyte subclusters would significantly improve our understanding of the underlying cellular events contributing to OA progression.
Researchers also identified abnormal populations in OA. Swahn et al. [102] discovered a subset of genes associated with senescence that expanded in OA. Through cellular interaction analysis using CellChat, it was found to play an essential role in connecting with other cartilage clusters via ECM, TNC, and TGF-β signaling pathways [102]. The dysregulated gene FAP in this cluster was shown to contribute to OA pathogenesis, while upregulated ZEB1 contributed to chondrocyte senescence [102]. Wijesinghe et al. [147] identified an obesity-specific subset characterized by an inflammatory endotype. Lv et al. [148] revealed a chondrocyte cluster expressing ferroptotic hallmarks and genes preferentially. Gene set variation analysis demonstrated that TRPV1 promoted GPX4 expression to regulate chondrocyte ferroptosis, which was verified in Gpx4+/− mice [148]. However, Wen et al. [149] discovered that although senescent chondrocytes hyperactivate ferroptosis, they may overexpress Solute carrier eamily 1 member 3 (SLC1A3, also known as EAAT1) to activate the glutathione system as a countermeasure against ferroptosis through metabolomics analysis. These studies suggest a close relationship between chondrocyte senescence and ferroptosis. Qu et al. [150] revealed a SPP1+ chondrocyte cluster exhibiting stronger angiogenic capacity and aging characteristics. Obeidat et al. [151] identified a subcluster of nociceptors co-expressing Piezo2 and Ntrk1 which is highly relevant to pain in OA (Fig. 5b). Synovitis, a common feature of OA involving active fibroblasts [139], was investigated by Nanus et al. who deciphered fibroblasts from different stages of OA at single-cell resolution [152]. Functional pathway analysis revealed that fibroblast subsets from painful sites promoted fibrosis, inflammation, and neuronal growth [152]. Knights et al. [153] found that Wnt/β-catenin signaling was overactive in post-traumatic OA (PTOA) synovium, with Rspo2 strongly induced after injury and secreted exclusively by Prg4hi lining fibroblasts, further increasing pathological crosstalk and contributing to the inflammation in PTOA. Researchers are also working on elucidating the role of immune cells in the OA [110, 154,155,156,157,158,159,160]. Lu et al. [159] revealed IGF2BP3 as a potential macrophage mediator in silico, and verified its function of promoting macrophage M1 polarization and inflammation in vitro. Sebastian et al. [154] identified a macrophage population enriched for phagocytic genes and growth factors. Meanwhile, IL-1β could promote GDF15 expression in OA chondrocytes and induce a senescence phenotype [160] (Fig. 5c). These findings have significant implications for advancing research initiatives aimed at developing customized treatments to address specific pathological populations in OA.
RA is another common joint disease characterized by synovial membrane inflammation, leukocyte infiltration, and aggressive fibroblasts [4, 161, 162]. Single-cell RNA analysis unveiled the intricate roles of immune cells, fibroblasts, and fibroblast-like synoviocyte subclusters in driving inflammation, bone erosion, and other pathological processes in rheumatoid arthritis (Fig. 6). Orange et al. [163] discovered circulating CD45−CD31−PDPN+ preinflammatory mesenchymal cells that expand before an RA flare but decrease during exacerbation. Jonsson et al. [164] revealed that fluid CD8+ T cells in synovial tissue belong to an effector CD8+ T cell population with high expression of granzyme K and low expression of granzyme B and perforin. These cells were found to be major cytokine producers with low cytotoxic potential [164]. Argyriou et al. [165] identified 2 peripheral helper T cell states and a cytotoxic CD4+ T cell subset with a common differentiation pathway in the synovial fluid of RA patients at single-cell resolution. Besides, SIGIRR, preferentially expressed by memory CD4+ T cells, could inversely regulate RA disease activity via IL-1/C/EBPβ/TNF-α signaling axis [166]. It can be concluded that T cells play an essential role in the pathological mechanisms of RA. Moreover, ACPA+ and RF+ B cells were more abundant in the peripheral blood of RA patients and exhibited distinct transcriptional programs, implying 2 different molecular mechanisms that contribute to the increased inflammation in RA [167]. Meednu et al. [168] identified an NR4A+ synovial B cell population that co-expresses lymphotoxins α, β, and IL-6 and functions in ectopic lymphoid aggregation (Fig. 6a).
Myeloid cells are also tightly involved in the development of RAs [26, 169,170,171,172,173] (Fig. 6a). Alivernini et al. [169] identified two types of synovial tissue macrophages (MerTKposTREM2high and MerTKposLYVE1pos) enriched with negative regulators of inflammation, whose potential to induce remission in RA was confirmed by their ability to elicit a reparative response in synovial fibroblasts. Conversely, HBEGF+ inflammatory macrophages were identified in the synovium, promoting fibroblast invasiveness through an epidermal growth factor receptor-dependent manner [170]. Zhang et al. [26] identified a cluster of proinflammatory monocytes as the major source of IL1B production. In addition, Zhang et al. [173] discovered a novel RANK+ TLR2− monocyte population that negatively regulates osteoclast fusion. Although these cells can differentiate into a TRAP+ osteoclast lineage, they fail to undergo fusion and form osteoclasts [173] (Fig. 6a). Therefore, myeloid cells also serve as a trigger for the occurrence and development of RA.

Single-cell RNA analysis unveiled the intricate roles of immune cells, fibroblasts, and fibroblast-like synoviocyte subclusters in driving inflammation, bone erosion, and other pathological processes in rheumatoid arthritis. a Effect CD8+ T cells are the major cytokine producer in RA. IL1 activates C/EBPβ phosphorylation to promote TNF production in Memory CD4+ T cells while SIGIRR plays an inhibitory role. Ectopic lymphoid B cells are activated in RA synovium. Myeloid cells play an important role in joint repairment and destruction. b Fibroblasts promote joint inflammation in RA, and FLSs drives long-term RA and contribute to bone erosion. RA rheumatoid arthritis, pC/EBPβ phosphorylated CCAAT/enhancer binding protein β, DUSP1 dual specificity protein phosphatase 1, KLF2 krueppel-like factor 2, EGF epidermal growth factor, THY1 thy-1 membrane glycoprotein, TNF tumor necrosis factor, TGF-β transforming growth factor-β, FGF10 fibroblast growth factor 10, FLS fibroblast-like synoviocyte, IL interleukin, JAG1 protein jagged-1, NOTCH3 notch homolog protein 3, CD cluster of differentiation, NR4A1 nuclear receptor subfamily 4 immunity group A member 1, RANK receptor activator for nuclear factor-κB, TLR2 Toll-like receptor 2, FAP fibroblast activation protein, proNGF nerve growth factor precursor, p75NTR p75 neurotrophin receptor, SIGIRR single immunoglobulin IL-1R-related receptor
Aggressive fibroblasts also play an important role in the progression of RA (Fig. 6b). Wei et al. [174] found that neurogenic locus notch homolog protein 3 (NOTCH3) signaling from vascular endothelial cells drives transcriptional and spatial gradients in fibroblasts. In mice, blocking NOTCH3 signaling attenuated inflammation and prevented joint damage in RA. Croft et al. [175] classified FAPα+THY1+ immune effector fibroblasts and FAPα+THY1− bone destructive fibroblasts, while Zhang et al. [26] identified THY1(CD90)+HLA-DRAhigh subliming fibroblasts as the potential major source of IL-6 in the synovium of RA patients. Chen et al. [176] identified HBEGF+ fibroblasts as being related to RA remission, improving our understanding of RA pathogenesis and treatment research. Farina et al. [177] discovered that the active proNGF/p75NTR axis could promote pro-inflammatory responses in synovial fibroblasts and further contribute to chronic synovial inflammation. scRNA-seq also revealed that fibroblasts are susceptible to ferroptosis, but TNF signaling promotes cystine uptake and glutathione biosynthesis to protect them from ferroptosis, as validated in vitro [178]. Thus, fibroblasts primarily contribute to RA pathology through inducing inflammation. In addition, fibroblast-like synoviocytes (FLS) are extensively involved in the mechanism underlying RA development. Smith et al. [179] found that myeloid and T cell-derived cytokines, such as TNF, IFN-γ, and IL-1β can drive 4 distinct states of FLS. Biesemann et al. [180] revealed that FLS serves as a receptor for TNF and IL-6 in OA, and the combination of TNF and IL-6 antibodies can result in sustained long-term remission in mouse models. In addition, in relapse RA patients, scRNA-seq showed that the fibroblast growth factor pathway was highly activated in lining FLS subsets and associated with bone erosions [181]. The importance of the fibroblast growth factor pathway in relapsed RA has been verified in vitro experiments and RA animal models, providing valuable insights into treatment [181] (Fig. 6b). These studies illustrated the altered function of fibroblasts, improving our understanding of the cellular basis of OA.
scRNA-seq provides a promising approach for investigating joint health and disease. However, difficulties in dissociating articular tissue such as the posterior synovium, make it challenging to identify specific pathological changes that may play critical roles in particular diseases. Therefore, it is necessary to develop more precise methods for tissue separation or utilize spatial transcriptomic analysis with single-cell accuracy.
The roles of stem cells in bone regeneration and disease
Skeletal stem/progenitor cells are critical for maintaining the homeostatic microenvironment of bones. In 2014, Zhou et al. [182] uncovered LepR+ skeletal stem cells (SSCs) in the bone marrow. Moreover, Mo et al. [25] used scRNA-seq to characterize the cellular heterogeneity in LepR+ SSCs, discovering a quiescent Notch3+ subcluster associated with the vasculatures and osteo-chondrogenic differentiation via Monocle2 analysis, as well as a Sca1+ subcluster with high clonogenic activity. Chan et al. [183] and Worthley et al. [184] identified self-renewal and multipotent SSCs from the growth plate of newborn mice. Subsequently, SSCs were also found within the periosteum of postnatal long bones and calvaria [185]. Correspondingly, Chan et al. [186] identified human SSCs in the growth plate of 17-week-old fetal long bones. He et al. [22] further explored the emergence and features of human embryonic SSCs during early bone formation. By comparing human limb buds at 5 weeks post conception (WPC) and long bones at 8 WPC, they discovered 16 clusters, including PRRX1+ limb bud mesenchymal subsets and osteo-chondrogenic progenitors with differentiation potential into osteogenic and chondrogenic lineages [22]. Ambrosi et al. [187] revealed that the decline of SSCs in aged mice was connected with diminished transcriptomic diversity, which could be reversed by a combination treatment of BMP2 and the CSF1 antagonist. Yin et al. [188] revealed a Scx+Hoxd13+ musculoskeletal stem cell population through scRNA-seq analysis on E10.5, E12.5, and E15.5 murine limbs, which is indispensable for bone development. Meanwhile, Hao et al. [189] analyzed mouse hindlimb buds, postnatal long bones, and fractured long bones at single-cell resolution, and identified Cd168+ skeletal stem/progenitor cells (SSPCs) with highly replicating capacity and osteochondrogenic potential in embryonic and postnatal long bones. Additionally, Sivaraj et al. [80] investigated the differences between bone marrow stromal cells (BMSCs) from the metaphysis and diaphysis and demonstrated the regulatory effect of PDGFR-β signaling and the transcription factor Jun-B on BMSCs fates. The bone marrow microenvironment also plays a crucial role in regulating hematopoiesis [190]. Tikhonova et al. [38] revealed cellular heterogeneity within the bone marrow niche using scRNA-seq under stress conditions and showed an adipocytic skewing of perivascular cells. Baccin et al. [21] identified 2 Cxcl12-abundant-reticular (CAR) cell subsets (Adipo-CAR and Osteo-CAR) localized to sinusoidal and arteriolar surfaces, respectively. These subsets act as potential cytokine-secreting clusters and maintain the perivascular microenvironment.
Fractures are the most common type of bone injury, and immune cells actively participate in the process of fracture healing [191]. Zhang et al. [57] analyzed fresh and aged fracture bones of mice by scRNA-seq and identified 13 clusters. Among these clusters, B cells exhibited significant variations. Moreover, progenitor cells are essential in fracture repair. Julien et al. [192] demonstrated that skeletal muscle progenitors adopt a fibrogenic fate before engaging in chondrogenesis following a fracture. They subsequently integrated the dataset from healthy and fractured mice for 3 d post-injury, revealing a cluster of fibrochondro progenitor (FCP) primarily located at the periosteum in injured tissues [193]. Pseudotime analysis using Monocle3 uncovered FCP processing with both fibrotic and chondrogenic trajectories, highlighting the important role of FCP in fracture repair [193]. Serowoky et al. [194] found that deficient Hedgehog signaling probably leads to failed recruitment of Cxcl12-expressing SSPCs, emphasizing the importance of Shh in large-scale bone regeneration. Cxcl12+ BMSCs undergo identity conversion into a skeletal stem cell-like state in response to injury, which was associated with the Wnt signaling pathway [195]. On the other hand, skeletal muscle mesenchymal progenitors adopt a fibrogenic fate before engaging in chondrogenesis after fracture, which elucidates the central role played by skeletal muscle in bone regeneration [192]. Additionally, fractures can be complications arising from osteoporosis, particularly among elderly individuals [5]. Abnormal activation of osteoclasts has been closely associated with osteoporosis, and Gingival tissue-derived MSCs (GMSCs) have been found to inhibit osteoclast activity [196, 197]. Based on this finding, Wu et al. conducted scRNA-seq analysis of GMSCs and identified a CD39+ subcluster that specifically expressed osteogenic genes such as BMP2 and RUNX2, exerting its osteogenic capacity via the Wnt/β-catenin pathway [198]. These studies provide novel insights into the mechanism of fractures and potential therapeutic targets.
Osteosarcoma (OS) is one of the most common bone malignancies worldwide, with an estimated global incidence rate of approximately 4.8/1,000,000 [199]. Guo et al. [200] found a cluster of osteosarcoma cells exhibiting highly expressing levels of collagen type VI alpha 1 chain (COL6A1), collagen type VI α 3 chain (COL6A3), and MIF, which were closely associated with lung metastasis. In recurrent OS cases, a subcluster of cancer-associated fibroblasts exhibited increased infiltration and enrichment in the epithelial-mesenchymal transition pathway [201]. Apart from OS, giant cell tumor of bone (GCTB) represents another common bone tumor that rarely leads to mortality but significantly elevates the risk of bone fractures and even disability [202]. The presence of osteoclasts and other immune cells in the tumor microenvironment plays a crucial role in these complications. Zhou et al. [203] used scRNA-seq to compare the transcriptome profiles between primary tumors, recurrent lesions, and pulmonary metastatic sites in osteosarcoma patients. They found a distinct cluster consisting of FABP4+ macrophages infiltrating in lung metastatic osteosarcoma lesions while observing heightened infiltration levels of osteoclast across all patients [203]. Feng et al. [204] characterized major clusters of macrophages, osteoclasts, and NK/T cells from GCTB patients and resolved the intracellular communication networks of immune cells via CellPhoneDB analysis, highlighting the role of RANK-RANKL signaling in inducing migration of osteoclasts to osteoblasts.
Despite the exceptional resolution provided by scRNA-seq, which greatly enhances our understanding of skeletal biology, certain limitations persist. Bones are primarily composed of multiple lamellar layers, resulting in a highly dense tissue structure, which may impede the effectiveness of scRNA-seq in capturing cells if the matrix is not fully disassembled. As a result, it is imperative to employ proper grinding methods and allocate sufficient time for enzymatic hydrolysis before cell separation to ensure precise and reliable outcomes.
Regeneration of muscle and tendon in skeletal disorders
Skeletal muscle regeneration following injury relies on microsatellite cells, also known as muscle stem cells, to restore the muscular microenvironment [205]. Giordani et al. [206] depicted the cellular landscape of adult mouse hindlimb muscles and demonstrated that Scx+ cell clusters give rise to tenocytes, while Itga7+Vcam1− cell clusters exhibit myogenic potential and enhance muscle stem cell (MuSC) engraftment following transplantation [206]. Andre portrayed the atlas of injured muscle and identified a novel cluster of satellite cells that might function as sensors for muscle infection or injury via the antiviral interferon pathway [207]. These atlas studies have enhanced the comprehensive understanding of the muscular ecosystem during hemostasis and repair. Penaloza et al. [208] revealed the heterogeneity of Mesp1+ lineage cells, which contribute to cardiac, hematopoietic, and skeletal myogenic development, and demonstrated potential differential trajectories in single-cell resolution. Moreover, Yang et al. [209] discovered a Pax3-expressing melanocyte population with robust myogenic potential, which was induced from the skin by a novel small-molecule cocktail. Although myosatellite cells are indispensable for muscle regeneration, it is also dependent on the crosstalk between MuSCs and components within their niches [210]. De Micheli et al. [211] analyzed the scRNA-seq data from hindlimb muscles of myotoxin-induced models and found that FGF2, TGF-β1, and RSPO3 regulate proliferation of myogenic stem/progenitor cell through a Syndecan-dependent mechanism. This proposed interaction network suggests a potential role for Syndecans in regulating myogenic differentiation [211]. In addition, Xi et al. [212] employed scRNA-seq to delineate the “roadmap” of human skeletal muscle and revealed the co-regulated gene networks and transcription factors that are present at distinct myogenic stages. Guo et al. [213] discovered that SRSF2 is a key regulator governing the entry of Myf5 cells into the myogenic program, ensuring their survival by preventing premature differentiation and apoptosis. Scott et al. [214] unveiled that HIC1 regulates tissue-resident mesenchymal progenitors to maintain quiescence and facilitate muscle regeneration. Epigenetically, circular RNAs also exert significant influence on muscle regeneration, as demonstrated by Yan et al. [215], who revealed that circFgfr2 regulates myogenesis and muscle regeneration through the activation of the JNK/MAPK pathway across 27 developmental stages in pig skeletal muscle.
Similar to muscle, tendons also possess the capacity to undergo self-repair. Given the importance of regulating tendon differentiation for effective self-repair, Kaji et al. [216] established directed differentiation models based on developmental cues and scRNA-seq analysis. These models successfully generated tendon and fibrocartilage cells from mouse embryonic stem cells by activating TGF-β and hedgehog pathways. They also identified retinoic acid signaling as a critical regulator of the cell fate switch between TGF-β-induced tendon and fibrocartilage lineages [216]. Moreover, tissue-resident tendon stem cells are also indispensable for tendon repair. Harvay et al. [217] revealed a cluster of Tppp3+ cells as the potential tendon stem cells, which were shown to generate new tenocytes and self-renew upon injury through lineage tracing. Fan et al. [218] found Cd9+Cd271+ tendon stem/progenitor cells characterized by nerve growth factor secretion primarily involved in the conversion from neonate to adult tendon. Fang et al. [101] demonstrated the clonogenicity and multipotency of Gli1-expressing progenitors, which function as stem cells during tendon regeneration, while Harvey et al. [217] found that PDGFRA-expressing Tppp3+ tendon stem cells were regulated by PDGFR-AA to produce new tenocytes. By combining single-cell gene regulatory network analysis, in vitro inhibitor identification, and in vivo deletion of specific genes related to tendons, Fan et al. [218] verified that the SHP2 signaling pathway is a crucial regulator for tendon maturation. Furthermore, the tendon microenvironment plays a crucial role in influencing tendon repair. Muscat et al. [219] identified the presence of macrophages and T cells in adult tendons using scRNA-seq, which contributes to the homeostasis in tendons. Following the knockout of chemokine C-C-motif receptor 2 (CCR2), a key molecule for macrophage recruitment, there was an observed decrease in myofibroblast and impaired functional recovery during the later stages of healing [219].
Challenges and prospects
In the developing field of musculoskeletal research, the advent of scRNA-seq has ushered in a new era, enabling researchers to delve into the intricacies of the transcriptome with unprecedented precision at the single-cell resolution. This powerful tool has made significant contribution to elucidating the underlying mechanism of various diseases. Nevertheless, scRNA-seq faces several challenges that must be addressed when applied in skeletal system research.
Firstly, the sheer volume of data generated by scRNA-seq poses a formidable challenge, as it produces vast amounts of high-dimensional data that complicates the extraction of information with biological importance. Advanced bioinformatic analysis can help pinpoint key factors amidst the data explosion. For example, Cell BLAST, developed by Cao et al. [220], empowers the precise and swift retrieval and annotation of newly generated single-cell data within existing databases, thereby enhancing the overall accuracy and efficiency of the annotation process. To tackle the challenge posed by high-dimensional data processing, it becomes imperative to delve deeper into the intricacies of this issue. The future may witness the development of more sophisticated machine learning algorithms for constructing more accurate prediction models and innovative dimensionality reduction techniques to select more vital factors for analysis. These developments will empower researchers to extract biologically significant information from complex datasets more efficiently. It is essential to recognize that these findings must be verified by rigorous biological experiments to clarify their biological validity and clinical relevance.
Secondly, the lack of comparability among study outcomes in skeletal research arises from the heterogeneity in sampling standards, sequencing methods, and analysis approaches that have been widely employed in scRNA-seq. Moreover, difficulties in data integration due to negative data-sharing behaviors greatly diminish the value of resources in studies. This challenge can be attributed to the absence of standardized practices across laboratories. To overcome this obstacle, future endeavors should focus on establishing clear standards and specifications that ensure data consistency across different research settings. Initiatives aimed at creating universally accepted standardization processes and fostering shared data principles are essential milestones for advancing the field. Encouraging multi-center collaborations emerges as another paramount strategy to enhance the reliability of scRNA-seq applications in skeletal research. By promoting a collective commitment to standardized practices, researchers can not only ensure the reproducibility of their findings but also facilitate robust data sharing. Such collaborative efforts are pivotal for realizing the full potential of scRNA-seq by enabling more reliable, comparable, and impactful results that transcend individual research boundaries. Ultimately, the establishment of a shared framework will amplify the collective impact of research endeavors and accelerate progress in musculoskeletal studies.
In addition to scRNA-seq, a plethora of emerging single-cell sequencing technologies are poised to revolutionize our understanding of skeletal research, offering more nuanced insights into cellular dynamics and molecular mechanisms. Single-cell ATAC-seq, for instance, presents a groundbreaking approach by unraveling chromatin accessibility profiles at the single-cell level [221]. The approach that uses transposase to capture chromatin openness provides researchers with the opportunity to employ high-throughput sequencing for an in-depth examination of chromatin accessibility and epigenetic properties [221]. Not only does this technique provide intricate details about the accessibility of genomic regions but also sheds light on the complex landscape of transcriptional regulation within individual cells. The ability to discern chromatin accessibility offers a valuable complement to scRNA-seq, enabling researchers to delve deeper into the epigenetic underpinnings of skeletal processes. Spatial transcriptome, another emerging frontier, addresses a critical limitation of scRNA-seq by restoring spatial context to gene expression patterns [222]. It employs either gene chips or image-based technologies to transform the gene expression information from sampled sites into digital signals [222]. By visualizing gene expression and distribution within tissue sections, spatial transcriptomics enables researchers to discern how cells interact in their native microenvironment. This capability provides a more holistic understanding of the spatial organization of cell populations within skeletal tissues. Multi-omics approaches that integrate genomics, metagenomics, transcriptomics, proteomics, and metabolomics offer a more comprehensive and insightful perspective on the pathological mechanism in orthopedic diseases [223]. Furthermore, scRNA-seq brings plenty of clues for the theoretical basis of utilizing genetic models and allows for the integration of clonal relationships into these molecular landscapes [224]. Sarah Bowling et al. [225] introduced the CRISPR array repair lineage tracing mouse line and accompanying analysis tools, which enable the simultaneous investigation of lineage and transcriptomic information in individual cells in vivo. A novel sequencing approach called Camellia-seq has recently emerged, allowing for the concurrent measurement of chromatin accessibility, DNA methylation, gene expression, and lineage information within individual cells [226]. Coupled with Cas9-TdT CRISPR array repair lineage tracing (DARLIN), Li et al. [226] designed a stable inducible lineage-labeling genetic mouse model capable of labeling approximately 1018 gene sites, and used Camellia-seq to systematically unveil unprecedented insights into the cellular fate decision process at the single-cell level. As we embrace these advancements, the combined application of scRNA-seq with these emerging technologies promises to unveil a more detailed and interconnected landscape of skeletal biology. These innovative tools not only complement the limitations of scRNA-seq but also pave the way for a more holistic and integrative exploration of the complexities inherent in musculoskeletal research.
Conclusions
In this review, we outlined the essential steps for acquiring high-quality single-cell suspensions from skeletal tissues, discussed the commonly employed scRNA-seq platforms in skeletal system research, and elucidated the indispensable bioinformatic analysis pipelines crucial for deciphering cellular heterogeneity and responses in skeletal homeostasis and diseases. These cutting-edge technologies hold promise in our pursuit of a comprehensive understanding and effective management of skeletal disorders. By harnessing the power of scRNA-seq technology, we can effectively address current challenges in skeletal research, enhance our understanding of the underlying mechanisms driving relevant diseases, propel precision medicine advancements in this field, and ultimately contribute to the prevention and treatment of skeletal disorders in military medicine.
Availability of data and materials
The datasets analyzed during the current study are available in the GEO repository (GSE160756).
Abbreviations
- AF:
-
Annulus fibrosus
- AS:
-
Ankylosing spondylitis
- BMSCs:
-
Bone marrow stromal cells
- CAR:
-
Cxcl12-abundant-reticular
- COL6A1:
-
Collagen type VI alpha 1 chain
- COL6A3:
-
Collagen type VI alpha 3 chain
- DARLIN:
-
Cas9-TdT CRISPR array repair lineage tracing
- DDD:
-
Degenerative disc disease
- DEGs:
-
Differentially expressed genes
- DR:
-
Dimensionality reduction
- ECM:
-
Extracellular matrix
- ECs:
-
Effector chondrocytes
- FACS:
-
Flow cytometry cell sorting
- FCP:
-
Fibrochondro progenitor
- FLS:
-
Fibroblast-like synoviocyte
- GCTB:
-
Giant cell tumor of bone
- GMSCs:
-
Gingival tissue-derived MSCs
- GRN:
-
Gene regulatory network
- GSEA:
-
Gene set enrichment analysis
- IVD:
-
Intervertebral disc
- MACS:
-
Magnetic bead sorting
- MIF:
-
Migration inhibitory factor
- MNN:
-
Mutual nearest neighbors
- MST:
-
Minimum spanning tree
- MuSC:
-
Muscle stem cell
- NAMPT:
-
Nicotinamide phosphoribosyltransferase
- NK:
-
Natural killer
- NOTCH3:
-
Neurogenic locus notch homolog protein 3
- NP:
-
Nucleus pulposus
- NPPC:
-
Nucleus pulposus progenitor cells
- OA:
-
Osteoarthritis
- PBMCs:
-
Peripheral blood mononuclear cells
- ProNPs:
-
NP progenitors
- PTN:
-
Pleiotrophin
- QC:
-
Quality control
- RA:
-
Rheumatoid arthritis
- SCENIC:
-
Single-cell regulatory network inference and clustering
- scRNA-seq:
-
Single-cell RNA sequencing
- SPP1:
-
Secreted phosphoprotein 1
- SSCs:
-
Skeletal stem cells
- SSPCs:
-
Skeletal stem/progenitor cells
- TGF-β:
-
Transforming growth factor-β
- TNC:
-
Tenascin-C
- t-SNE:
-
t-distributed stochastic neighbor embedding
- UMAP:
-
Uniform manifold approximation and projection
- WPC:
-
Weeks post conception
References
Zhou M, Wang H, Zeng X, Yin P, Zhu J, Chen W, et al. Mortality, morbidity, and risk factors in China and its provinces, 1990–2017: a systematic analysis for the global burden of Disease Study 2017. Lancet. 2019;394(10204):1145–58.
Knezevic NN, Candido KD, Vlaeyen JWS, Van Zundert J, Cohen SP. Low back pain. Lancet. 2021;398(10294):78–92.
Hunter DJ, Bierma-Zeinstra S, Osteoarthritis. Lancet. 2019;393(10182):1745–59.
Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. 2016;388(10055):2023–38.
Compston JE, McClung MR, Leslie WD. Osteoporos Lancet. 2019;393(10169):364–76.
Yassi A. Repetitive strain injuries. Lancet. 1997;349(9056):943–7.
Boskey AL. Musculoskeletal disorders and orthopedic conditions. JAMA. 2001;285(5):619–23.
Zhang Z, Zhou J, Liu C, Zhang J, Shibata Y, Kong N, et al. Emerging biomimetic nanotechnology in orthopedic diseases: progress, challenges, and opportunities. Trends Chem. 2022;4(5):420–36.
Yang G, Wang Y, Zeng Y, Gao G, Liang X, Zhou M, et al. Rapid health transition in China, 1990–2010: findings from the global burden of Disease Study 2010. Lancet. 2013;381(9882):1987–2015.
Xu PC, Xuan M, Cheng B. Effects and mechanism of platelet-rich plasma on military drill injury: a review. Mil Med Res. 2020;7(1):56.
Zhang Y, Wang J, Yu C, Xia K, Yang B, Zhang Y, et al. Advances in single-cell sequencing and its application to musculoskeletal system research. Cell Prolif. 2022;55(1):e13161.
Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods. 2009;6(5):377–82.
Owens B. Genomics: the single life. Nature. 2012;491(7422):27–9.
Stuart T, Satija R. Integrative single-cell analysis. Nat Rev Genet. 2019;20(5):257–72.
Kiselev VY, Andrews TS, Hemberg M. Challenges in unsupervised clustering of single-cell RNA-seq data. Nat Rev Genet. 2019;20(5):273–82.
Su M, Pan T, Chen QZ, Zhou WW, Gong Y, Xu G, et al. Data analysis guidelines for single-cell RNA-seq in biomedical studies and clinical applications. Mil Med Res. 2022;9(1):68.
Islam S, Zeisel A, Joost S, La Manno G, Zajac P, Kasper M, et al. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat Methods. 2014;11(2):163–6.
Shapiro E, Biezuner T, Linnarsson S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat Rev Genet. 2013;14(9):618–30.
Culemann S, Grüneboom A, Nicolás-Ávila JÁ, Weidner D, Lämmle KF, Rothe T, et al. Locally renewing resident synovial macrophages provide a protective barrier for the joint. Nature. 2019;572(7771):670–5.
Salhotra A, Shah HN, Levi B, Longaker MT. Mechanisms of bone development and repair. Nat Rev Mol Cell Biol. 2020;21(11):696–711.
Baccin C, Al-Sabah J, Velten L, Helbling PM, Grünschläger F, Hernández-Malmierca P, et al. Combined single-cell and spatial transcriptomics reveal the molecular, cellular and spatial bone marrow niche organization. Nat Cell Biol. 2020;22(1):38–48.
He J, Yan J, Wang J, Zhao L, Xin Q, Zeng Y, et al. Dissecting human embryonic skeletal stem cell ontogeny by single-cell transcriptomic and functional analyses. Cell Res. 2021;31(7):742–57.
Gan Y, He J, Zhu J, Xu Z, Wang Z, Yan J, et al. Spatially defined single-cell transcriptional profiling characterizes diverse chondrocyte subtypes and nucleus pulposus progenitors in human intervertebral discs. Bone Res. 2021;9(1):37.
Bian Q, Cheng YH, Wilson JP, Su EY, Kim DW, Wang H, et al. A single cell transcriptional atlas of early synovial joint development. Development. 2020;147(14):dev185777.
Mo C, Guo J, Qin J, Zhang X, Sun Y, Wei H, et al. Single-cell transcriptomics of LepR-positive skeletal cells reveals heterogeneous stress-dependent stem and progenitor pools. EMBO J. 2022;41(4):e108415.
Zhang F, Wei K, Slowikowski K, Fonseka CY, Rao DA, Kelly S, et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat Immunol. 2019;20(7):928–42.
Tu J, Li W, Yang S, Yang P, Yan Q, Wang S, et al. Single-cell transcriptome profiling reveals multicellular ecosystem of nucleus pulposus during degeneration progression. Adv Sci. 2022;9(3):e2103631.
Ji Q, Zheng Y, Zhang G, Hu Y, Fan X, Hou Y, et al. Single-cell RNA-seq analysis reveals the progression of human osteoarthritis. Ann Rheum Dis. 2019;78(1):100–10.
Li PH, Kong XY, He YZ, Liu Y, Peng X, Li ZH, et al. Recent developments in application of single-cell RNA sequencing in the tumour immune microenvironment and cancer therapy. Mil Med Res. 2022;9(1):52.
Greenblatt MB, Ono N, Ayturk UM, Debnath S, Lalani S. The unmixing problem: a guide to applying single-cell RNA sequencing to bone. J Bone Min Res. 2019;34(7):1207–19.
Gu Y, Hu Y, Zhang H, Wang S, Xu K, Su J. Single-cell RNA sequencing in osteoarthritis. Cell Prolif. 2023;56(12):e13517.
Reichard A, Asosingh K. Best practices for preparing a single cell suspension from solid tissues for flow cytometry. Cytometry A. 2019;95(2):219–26.
Sutermaster BA, Darling EM. Considerations for high-yield, high-throughput cell enrichment: fluorescence versus magnetic sorting. Sci Rep. 2019;9(1):227.
Yuan C, Pan Z, Zhao K, Li J, Sheng Z, Yao X, et al. Classification of four distinct osteoarthritis subtypes with a knee joint tissue transcriptome atlas. Bone Res. 2020;8(1):38.
Gleitz HFE, Snoeren IAM, Fuchs SNR, Leimkühler NB, Schneider RK. Isolation of human bone marrow stromal cells from bone marrow biopsies for single-cell RNA sequencing. STAR Protoc. 2021;2(2):100538.
Gulati GS, Murphy MP, Marecic O, Lopez M, Brewer RE, Koepke LS, et al. Isolation and functional assessment of mouse skeletal stem cell lineage. Nat Protoc. 2018;13(6):1294–309.
Gao M, Guo P, Liu X, Zhang P, He Z, Wen L, et al. Systematic study of single-cell isolation from musculoskeletal tissues for single-sell sequencing. BMC Mol Cell Biol. 2022;23(1):32.
Tikhonova AN, Dolgalev I, Hu H, Sivaraj KK, Hoxha E, Cuesta-Domínguez Á, et al. The bone marrow microenvironment at single-cell resolution. Nature. 2019;569(7755):222–8.
Yan Y, Fu R, Liu C, Yang J, Li Q, Huang RL. Sequential enzymatic digestion of different cartilage tissues: a rapid and high-efficiency protocol for chondrocyte isolation, and its application in cartilage tissue engineering. Cartilage. 2021;13(2suppl):S1064–76.
Shahini A, Vydiam K, Choudhury D, Rajabian N, Nguyen T, Lei P, et al. Efficient and high yield isolation of myoblasts from skeletal muscle. Stem Cell Res. 2018;30:122–9.
Bhagwat N, Dulmage K, Pletcher CH, Wang L, DeMuth W, Sen M, et al. An integrated flow cytometry-based platform for isolation and molecular characterization of circulating tumor single cells and clusters. Sci Rep. 2018;8(1):5035.
Adan A, Alizada G, Kiraz Y, Baran Y, Nalbant A. Flow cytometry: basic principles and applications. Crit Rev Biotechnol. 2017;37(2):163–76.
Wang L, Verbruggen G, Almqvist KF, Elewaut D, Broddelez C, Veys EM. Flow cytometric analysis of the human articular chondrocyte phenotype in vitro. Osteoarthritis Cartilage. 2001;9(1):73–84.
Ryan K, Rose RE, Jones DR, Lopez PA. Sheath fluid impacts the depletion of cellular metabolites in cells afflicted by sorting induced cellular stress (SICS). Cytometry A. 2021;99(9):921–9.
Méndez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466(7308):829–34.
Wang Z, Li X, Yang J, Gong Y, Zhang H, Qiu X, et al. Single-cell RNA sequencing deconvolutes the in vivo heterogeneity of human bone marrow-derived mesenchymal stem cells. Int J Biol Sci. 2021;17(15):4192–206.
Shalek AK, Satija R, Shuga J, Trombetta JJ, Gennert D, Lu D, et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature. 2014;510(7505):363–9.
Zeng W, Jiang S, Kong X, El-Ali N, Ball AR, Ma CIH, et al. Single-nucleus RNA-seq of differentiating human myoblasts reveals the extent of fate heterogeneity. Nucleic Acids Res. 2016;44(21):e158.
Paik DT, Cho S, Tian L, Chang HY, Wu JC. Single-cell RNA sequencing in cardiovascular development, disease and medicine. Nat Rev Cardiol. 2020;17(8):457–73.
Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161(5):1202–14.
Gao C, Zhang M, Chen L. The comparison of two single-cell sequencing platforms: BD rhapsody and 10x genomics chromium. Curr Genomics. 2020;21(8):602–9.
Mazid MA, Ward C, Luo Z, Liu C, Li Y, Lai Y, et al. Rolling back human pluripotent stem cells to an eight-cell embryo-like stage. Nature. 2022;605(7909):315–24.
Ding J, Adiconis X, Simmons SK, Kowalczyk MS, Hession CC, Marjanovic ND, et al. Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nat Biotechnol. 2020;38(6):737–46.
Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8:14049.
Huang ZY, Luo ZY, Cai YR, Chou CH, Yao ML, Pei FX, et al. Single cell transcriptomics in human osteoarthritis synovium and in silico deconvoluted bulk RNA sequencing. Osteoarthritis Cartilage. 2022;30(3):475–80.
Liu Y, Feng W, Dai Y, Bao M, Yuan Z, He M, et al. Single-cell transcriptomics reveals the complexity of the tumor microenvironment of treatment-naive osteosarcoma. Front Oncol. 2021;11:709210.
Zhang H, Wang R, Wang G, Zhang B, Wang C, Li D, et al. Single-cell RNA sequencing reveals B cells are important regulators in fracture healing. Front Endocrinol (Lausanne). 2021;12:666140.
Zhang X, Li T, Liu F, Chen Y, Yao J, Li Z, et al. Comparative analysis of droplet-based ultra-high-throughput single-cell RNA-seq systems. Mol Cell. 2019;73(1):130–42.e5.
Mereu E, Lafzi A, Moutinho C, Ziegenhain C, McCarthy DJ, Alvarez-Varela A, et al. Benchmarking single-cell RNA-sequencing protocols for cell atlas projects. Nat Biotechnol. 2020;38(6):747–55.
Hashimshony T, Wagner F, Sher N, Yanai I. CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification. Cell Rep. 2012;2(3):666–73.
Hedlund E, Deng Q. Single-cell RNA sequencing: technical advancements and biological applications. Mol Aspects Med. 2018;59:36–46.
Mizoguchi F, Slowikowski K, Wei K, Marshall JL, Rao DA, Chang SK, et al. Functionally distinct disease-associated fibroblast subsets in rheumatoid arthritis. Nat Commun. 2018;9(1):789.
Picelli S, Bjorklund AK, Faridani OR, Sagasser S, Winberg G, Sandberg R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods. 2013;10(11):1096–8.
Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36(5):411–20.
Wolf FA, Angerer P, Theis FJ. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19(1):15.
Balzer MS, Ma Z, Zhou J, Abedini A, Susztak K. How to get started with single cell RNA sequencing data analysis. J Am Soc Nephrol. 2021;32(6):1279–92.
Hwang B, Lee JH, Bang D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp Mol Med. 2018;50(8):1–14.
McGinnis CS, Murrow LM, Gartner ZJ. DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 2019;8(4):329–37.e4.
DePasquale EAK, Schnell DJ, Van Camp PJ, Valiente-Alandí Í, Blaxall BC, Grimes HL, et al. DoubletDecon: deconvoluting doublets from single-cell RNA-sequencing data. Cell Rep. 2019;29(6):1718–27.e8.
Wolock SL, Lopez R, Klein AM. Scrublet: computational identification of cell doublets in single-cell transcriptomic data. Cell Syst. 2019;8(4):281–91.e9.
Xi NM, Li JJ. Benchmarking computational doublet-detection methods for single-cell RNA sequencing data. Cell Syst. 2021;12(2):176–94.e6.
Luecken MD, Büttner M, Chaichoompu K, Danese A, Interlandi M, Mueller MF, et al. Benchmarking atlas-level data integration in single-cell genomics. Nat Methods. 2022;19(1):41–50.
Tran HTN, Ang KS, Chevrier M, Zhang X, Lee NYS, Goh M, et al. A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biol. 2020;21(1):12.
Haghverdi L, Lun ATL, Morgan MD, Marioni JC. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat Biotechnol. 2018;36(5):421–7.
Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, et al. Comprehensive integration of single-cell data. Cell. 2019;177(7):1888–902.e21.
Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods. 2019;16(12):1289–96.
Lotfollahi M, Wolf FA, Theis FJ. scGen predicts single-cell perturbation responses. Nat Methods. 2019;16(8):715–21.
van der Maaten L, Hinton G. Visualizing data using t-SNE. J Mach Learn Res. 2008;9(86):2579–605.
Becht E, McInnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol. 2018. https://doi.org/10.1038/nbt.4314. Online ahead of print.
Sivaraj KK, Jeong HW, Dharmalingam B, Zeuschner D, Adams S, Potente M, et al. Regional specialization and fate specification of bone stromal cells in skeletal development. Cell Rep. 2021;36(2):109352.
Al-Mossawi H, Yager N, Taylor CA, Lau E, Danielli S, de Wit J, et al. Context-specific regulation of surface and soluble IL7R expression by an autoimmune risk allele. Nat Commun. 2019;10(1):4575.
Cai S, Ming B, Ye C, Lin S, Hu P, Tang J, et al. Similar transition processes in synovial fibroblasts from rheumatoid arthritis and osteoarthritis: a single-cell study. J Immunol Res. 2019;2019:4080735.
Cribbs AP, Terlecki-Zaniewicz S, Philpott M, Baardman J, Ahern D, Lindow M, et al. Histone H3K27me3 demethylases regulate human Th17 cell development and effector functions by impacting on metabolism. Proc Natl Acad Sci U S A. 2020;117(11):6056–66.
Ayturk UM, Scollan JP, Goz Ayturk D, Suh ES, Vesprey A, Jacobsen CM, et al. Single-cell RNA sequencing of calvarial and long-bone endocortical cells. J Bone Min Res. 2020;35(10):1981–91.
Macqueen J. Some methods for classification and analysis of multivariate observations. Mathematics. 281 – 97; 1967.
Blondel VD, Guillaume JL, Lambiotte R, Lefebvre E. Fast unfolding of communities in large networks. J Stat Mech. 2008. https://doi.org/10.1088/1742-5468/2008/10/p10008.
Lancichinetti A, Fortunato S. Community detection algorithms: a comparative analysis. Phys Rev E Stat Nonlin Soft Matter Phys. 2009;80(5 Pt 2):056117.
Fortunato S, Barthelemy M. Resolution limit in community detection. Proc Natl Acad Sci U S A. 2007;104(1):36–41.
Amezquita RA, Lun ATL, Becht E, Carey VJ, Carpp LN, Geistlinger L, et al. Orchestrating single-cell analysis with Bioconductor. Nat Methods. 2020;17(2):137–45.
Traag VA, Waltman L, van Eck NJ. From Louvain to Leiden: guaranteeing well-connected communities. Sci Rep. 2019;9(1):5233.
Aran D, Looney A, Liu L, Wu E, Fong V, Hsu A, et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol. 2019;20(2):163–72.
Zhang A, O’Flanagan C, Chavez E, Lim J, Ceglia N, McPherson A, et al. Probabilistic cell-type assignment of single-cell RNA-seq for tumor microenvironment profiling. Nat Methods. 2019;16(10):1007–15.
Pliner H, Shendure J, Trapnell C. Supervised classification enables rapid annotation of cell atlases. Nat Methods. 2019;16(10):983–6.
Zhang Y, Zhang Y, Hu J, Zhang J, Guo F, Zhou M, et al. scTPA: a web tool for single-cell transcriptome analysis of pathway activation signatures. Bioinformatics. 2020;36(14):4217–9.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–9.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50.
Wang Q, Zhou C, Li X, Cai L, Zou J, Zhang D, et al. TGF-β1 promotes gap junctions formation in chondrocytes via Smad3/Smad4 signalling. Cell Prolif. 2019;52(2):e12544.
Gao B, Jiang B, Xing W, Xie Z, Luo Z, Zou W. Discovery and application of postnatal nucleus pulposus progenitors essential for intervertebral disc homeostasis and degeneration. Adv Sci (Weinh). 2022;9(13):e2104888.
Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, et al. SCENIC: single-cell regulatory network inference and clustering. Nat Methods. 2017;14(11):1083–6.
Fang F, Xiao Y, Zelzer E, Leong KW, Thomopoulos S. A mineralizing pool of Gli1-expressing progenitors builds the tendon enthesis and demonstrates therapeutic potential. Cell Stem Cell. 2022;29(12):1669–84.e6.
Swahn H, Li K, Duffy T, Olmer M, D’Lima DD, Mondala TS, et al. Senescent cell population with ZEB1 transcription factor as its main regulator promotes osteoarthritis in cartilage and meniscus. Ann Rheum Dis. 2023;82(3):403–15.
Fu W, Chen S, Yang R, Li C, Gao H, Li J, et al. Cellular features of localized microenvironments in human meniscal degeneration: a single-cell transcriptomic study. Elife. 2022;11:e79585.
Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014;32(4):381–6.
La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, et al. RNA velocity of single cells. Nature. 2018;560(7719):494–8.
Bergen V, Lange M, Peidli S, Wolf FA, Theis FJ. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat Biotechnol. 2020;38(12):1408–14.
Efremova M, Vento-Tormo M, Teichmann SA, Vento-Tormo R. CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat Protoc. 2020;15(4):1484–506.
Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan CH, et al. Inference and analysis of cell-cell communication using CellChat. Nat Commun. 2021;12(1):1088.
Wu X, Liu Y, Jin S, Wang M, Jiao Y, Yang B, et al. Single-cell sequencing of immune cells from anticitrullinated peptide antibody positive and negative rheumatoid arthritis. Nat Commun. 2021;12(1):4977.
Ling Z, Liu Y, Wang Z, Zhang Z, Chen B, Yang J, et al. Single-cell RNA-seq analysis reveals macrophage involved in the progression of human intervertebral disc degeneration. Front Cell Dev Biol. 2022;9:833420.
Hubaud A, Pourquié O. Signalling dynamics in vertebrate segmentation. Nat Rev Mol Cell Biol. 2014;15(11):709–21.
Oates AC, Morelli LG, Ares S. Patterning embryos with oscillations: structure, function and dynamics of the vertebrate segmentation clock. Development. 2012;139(4):625–39.
Diaz-Cuadros M, Wagner DE, Budjan C, Hubaud A, Tarazona OA, Donelly S, et al. In vitro characterization of the human segmentation clock. Nature. 2020;580(7801):113–8.
Li J, Wang L, Yu D, Hao J, Zhang L, Adeola AC, et al. Single-cell RNA sequencing reveals thoracolumbar vertebra heterogeneity and rib-genesis in pigs. Genomics Proteom Bioinf. 2021;19(3):423–36.
Yu H, Tang D, Wu H, Li C, Lu Y, He F, et al. Integrated single-cell analyses decode the developmental landscape of the human fetal spine. IScience. 2022;25(7):104679.
Zhou T, Chen Y, Liao Z, Zhang L, Su D, Li Z, et al. Spatiotemporal characterization of human early intervertebral disc formation at single-cell resolution. Adv Sci (Weinh). 2023;10(14):e2206296.
Wang H, Wang D, Luo B, Wang D, Jia H, Peng P, et al. Decoding the annulus fibrosus cell atlas by scRNA-seq to develop an inducible composite hydrogel: a novel strategy for disc reconstruction. Bioact Mater. 2022;14:350–63.
Han S, Zhang Y, Zhang X, Zhang H, Meng S, Kong M, et al. Single-cell RNA sequencing of the nucleus pulposus reveals chondrocyte differentiation and regulation in intervertebral disc degeneration. Front Cell Dev Biol. 2022;10:824771.
Lin P, Yan P, Zhu J, Huang S, Wang Z, Hu O, et al. Spatially multicellular variability of intervertebral disc degeneration by comparative single-cell analysis. Cell Prolif. 2023;56(10):e13464.
Li Z, Ye D, Dai L, Xu Y, Wu H, Luo W, et al. Single-cell RNA sequencing reveals the difference in human normal and degenerative nucleus pulposus tissue profiles and cellular interactions. Front Cell Dev Biol. 2022;10:910626.
Zhang Y, Han S, Kong M, Tu Q, Zhang L, Ma X. Single-cell RNA-seq analysis identifies unique chondrocyte subsets and reveals involvement of ferroptosis in human intervertebral disc degeneration. Osteoarthritis Cartilage. 2021;29(9):1324–34.
Zhou Z, Suo Y, Bai J, Lin F, Gao X, Shan H, et al. Matrix stiffness activating YAP/TEAD1-Cyclin B1 in nucleus pulposus cells promotes intervertebral disc degeneration. Aging Dis. 2023;14:1739–56.
Risbud MV, Shapiro IM. Role of cytokines in intervertebral disc degeneration: pain and disc content. Nat Rev Rheumatol. 2014;10(1):44–56.
Rohanifar M, Clayton SW, Easson GWD, Patil DS, Lee F, Jing L, et al. Single cell RNA-sequence analyses reveal uniquely expressed genes and heterogeneous immune cell involvement in the rat model of intervertebral disc degeneration. Appl Sci (Basel). 2022;12(16):8244.
Li W, Zhao Y, Wang Y, He Z, Zhang L, Yuan B, et al. Deciphering the sequential changes of monocytes/macrophages in the progression of IDD with longitudinal approach using single-cell transcriptome. Front Immunol. 2023;14:1090637.
Sun H, Wang H, Zhang W, Mao H, Li B. Single-cell RNA sequencing reveals resident progenitor and vascularization-associated cell subpopulations in rat annulus fibrosus. J Orthop Translat. 2022;38:256–67.
Li W, Zhang S, Zhao Y, Wang D, Shi Q, Ding Z, et al. Revealing the key MSCs niches and pathogenic genes in influencing CEP homeostasis: a conjoint analysis of single-cell and WGCNA. Front Immunol. 2022;13:933721.
Guo S, Yan M, Li X, Zhang S, Liu Z, Li K, et al. Single-cell RNA-seq analysis reveals that immune cells induce human nucleus pulposus ossification and degeneration. Front Immunol. 2023;14:1224627.
Braun J, Sieper J. Ankylosing spondylitis. Lancet. 2007;369(9570):1379–90.
Xu H, Yu H, Liu L, Wu H, Zhang C, Cai W, et al. Integrative single-cell RNA-seq and ATAC-seq analysis of peripheral mononuclear cells in patients with ankylosing spondylitis. Front Immunol. 2021;12:760381.
Ren C, Li M, Zheng Y, Cai B, Du W, Zhang H, et al. Single-cell RNA-seq reveals altered NK cell subsets and reduced levels of cytotoxic molecules in patients with ankylosing spondylitis. J Cell Mol Med. 2022;26(4):1071–82.
Karreman MC, Luime JJ, Hazes JMW, Weel AEAM. The prevalence and incidence of axial and peripheral spondyloarthritis in inflammatory bowel disease: a systematic review and meta-analysis. J Crohns Colitis. 2017;11(5):631–42.
Lefferts AR, Regner EH, Stahly A, O’Rourke B, Gerich ME, Fennimore BP, et al. Circulating mature granzyme B+ T cells distinguish Crohn’s disease-associated axial spondyloarthritis from axial spondyloarthritis and Crohn’s disease. Arthritis Res Ther. 2021;23(1):147.
Li Z, Chen S, Cui H, Li X, Chen D, Hao W, et al. Tenascin-C-mediated suppression of extracellular matrix adhesion force promotes entheseal new bone formation through activation of Hippo signalling in ankylosing spondylitis. Ann Rheum Dis. 2021;80(7):891–902.
Decker RS. Articular cartilage and joint development from embryogenesis to adulthood. Semin Cell Dev Biol. 2017;62:50–6.
Collins FL, Roelofs AJ, Symons RA, Kania K, Campbell E, Collie-Duguid ESR, et al. Taxonomy of fibroblasts and progenitors in the synovial joint at single-cell resolution. Ann Rheum Dis. 2023;82(3):428–37.
Gao M, Liu X, Guo P, Wang J, Li J, Wang W, et al. Deciphering postnatal limb development at single-cell resolution. IScience. 2023;26(1):105808.
Glyn-Jones S, Palmer AJR, Agricola R, Price AJ, Vincent TL, Weinans H, et al. Osteoarthr Lancet. 2015;386(9991):376–87.
Sebastian A, McCool JL, Hum NR, Murugesh DK, Wilson SP, Christiansen BA, et al. Single-cell RNA-seq reveals transcriptomic heterogeneity and post-traumatic osteoarthritis-associated early molecular changes in mouse articular chondrocytes. Cells. 2021;10(6):1462.
Li X, Liao Z, Deng Z, Chen N, Zhao L. Combining bulk and single-cell RNA-sequencing data to reveal gene expression pattern of chondrocytes in the osteoarthritic knee. Bioengineered. 2021;12(1):997–1007.
Wei K, Shu Z, Pu H, Xu H, Li S, Xiao J, et al. Cystathionine-γ-lyase attenuates inflammatory response and pain of osteoarthritis. Int Immunopharmacol. 2023;120:110289.
Chou CH, Jain V, Gibson J, Attarian DE, Haraden CA, Yohn CB, et al. Synovial cell cross-talk with cartilage plays a major role in the pathogenesis of osteoarthritis. Sci Rep. 2020;10(1):10868.
Wang X, Ning Y, Zhang P, Poulet B, Huang R, Gong Y, et al. Comparison of the major cell populations among osteoarthritis, Kashin-Beck disease and healthy chondrocytes by single-cell RNA-seq analysis. Cell Death Dis. 2021;12(6):551.
Li J, Fan C, Lv Z, Sun Z, Han J, Wang M, et al. Microtubule stabilization targeting regenerative chondrocyte cluster for cartilage regeneration. Theranostics. 2023;13(10):3480–96.
Hu X, Li Z, Ji M, Lin Y, Chen Y, Lu J. Identification of cellular heterogeneity and immunogenicity of chondrocytes via single-cell RNA sequencing technique in human osteoarthritis. Front Pharmacol. 2022;13:1004766.
Kang X, Zhang K, Wang Y, Zhao Y, Lu Y. Single-cell RNA sequencing analysis of human chondrocytes reveals cell-cell communication alterations mediated by interactive signaling pathways in osteoarthritis. Front Cell Dev Biol. 2023;11:1099287.
Wijesinghe SN, Badoume A, Nanus DE, Sharma-Oates A, Farah H, Certo M, et al. Obesity defined molecular endotypes in the synovium of patients with osteoarthritis provides a rationale for therapeutic targeting of fibroblast subsets. Clin Transl Med. 2023;13(4):e1232.
Lv Z, Han J, Li J, Guo H, Fei Y, Sun Z, et al. Single cell RNA-seq analysis identifies ferroptotic chondrocyte cluster and reveals TRPV1 as an anti-ferroptotic target in osteoarthritis. EBioMedicine. 2022;84:104258.
Wen Z, Xia G, Liang C, Wang X, Huang J, Zhang L, et al. Selective clearance of senescent chondrocytes in osteoarthritis by targeting excitatory amino acid transporter protein 1 to induce ferroptosis. Antioxid Redox Signal. 2023;39(4–6):262–77.
Qu Y, Wang Y, Wang S, Yu X, He Y, Lu R, et al. A comprehensive analysis of single-cell RNA transcriptome reveals unique SPP1+ chondrocytes in human osteoarthritis. Comput Biol Med. 2023;160:106926.
Obeidat AM, Wood MJ, Adamczyk NS, Ishihara S, Li J, Wang L, et al. Piezo2 expressing nociceptors mediate mechanical sensitization in experimental osteoarthritis. Nat Commun. 2023;14(1):2479.
Nanus DE, Badoume A, Wijesinghe SN, Halsey AM, Hurley P, Ahmed Z, et al. Synovial tissue from sites of joint pain in knee osteoarthritis patients exhibits a differential phenotype with distinct fibroblast subsets. EBioMedicine. 2021;72:103618.
Knights AJ, Farrell EC, Ellis OM, Lammlin L, Junginger LM, Rzeczycki PM, et al. Synovial fibroblasts assume distinct functional identities and secrete R-spondin 2 in osteoarthritis. Ann Rheum Dis. 2023;82(2):272–82.
Sebastian A, Hum NR, McCool JL, Wilson SP, Murugesh DK, Martin KA, et al. Single-cell RNA-seq reveals changes in immune landscape in post-traumatic osteoarthritis. Front Immunol. 2022;13:938075.
Zhao X, Younis S, Shi H, Hu S, Zia A, Wong HH, et al. RNA-seq characterization of histamine-releasing mast cells as potential therapeutic target of osteoarthritis. Clin Immunol. 2022;244:109117.
Gong Y, Yang J, Li X, Zhou C, Chen Y, Wang Z, et al. A systematic dissection of human primary osteoblasts in vivo at single-cell resolution. Aging. 2021;13(16):20629–50.
Liu W, Chen Y, Zeng G, Yang S, Yang T, Ma M, et al. Single-cell profiles of age-related osteoarthritis uncover underlying heterogeneity associated with disease progression. Front Mol Biosci. 2021;8:748360.
Zhou J, Zhao Z, He C, Gao F, Guo Y, Qu F, et al. Single-cell transcriptome analysis profile of meniscal tissue macrophages in human osteoarthritis. J Immunol Res. 2020;2020:8127281.
Lu Y, Zhang H, Pan H, Zhang Z, Zeng H, Xie H, et al. Expression pattern analysis of m6A regulators reveals IGF2BP3 as a key modulator in osteoarthritis synovial macrophages. J Transl Med. 2023;21(1):339.
Weng PW, Pikatan NW, Setiawan SA, Yadav VK, Fong IH, Hsu CH, et al. Role of GDF15/MAPK14 axis in chondrocyte senescence as a novel senomorphic agent in osteoarthritis. Int J Mol Sci. 2022;23(13):7043.
McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365(23):2205–19.
Mulder K, Patel AA, Kong WT, Piot C, Halitzki E, Dunsmore G, et al. Cross-tissue single-cell landscape of human monocytes and macrophages in health and disease. Immunity. 2021;54(8):1883–900.e5.
Orange DE, Yao V, Sawicka K, Fak J, Frank MO, Parveen S, et al. RNA identification of PRIME cells predicting rheumatoid arthritis flares. N Engl J Med. 2020;383(3):218–28.
Jonsson AH, Zhang F, Dunlap G, Gomez-Rivas E, Watts GFM, Faust HJ, et al. Granzyme K+ CD8 T cells form a core population in inflamed human tissue. Sci Transl Med. 2022;14(649):eabo0686.
Argyriou A, Wadsworth MH 2nd, Lendvai A, Christensen SM, Hensvold AH, Gerstner C, et al. Single cell sequencing identifies clonally expanded synovial CD4+ TPH cells expressing GPR56 in rheumatoid arthritis. Nat Commun. 2022;13(1):4046.
Teng X, Mou DC, Li HF, Jiao L, Wu SS, Pi JK, et al. SIGIRR deficiency contributes to CD4 T cell abnormalities by facilitating the IL1/C/EBPβ/TNF-α signaling axis in rheumatoid arthritis. Mol Med. 2022;28(1):135.
Lu DR, McDavid AN, Kongpachith S, Lingampalli N, Glanville J, Ju CH, et al. T cell-dependent affinity maturation and innate immune pathways differentially drive autoreactive B cell responses in rheumatoid arthritis. Arthritis Rheumatol. 2018;70(11):1732–44.
Meednu N, Rangel-Moreno J, Zhang F, Escalera-Rivera K, Corsiero E, Prediletto E, et al. Dynamic spectrum of ectopic lymphoid B cell activation and hypermutation in the RA synovium characterized by NR4A nuclear receptor expression. Cell Rep. 2022;39(5):110766.
Alivernini S, MacDonald L, Elmesmari A, Finlay S, Tolusso B, Gigante MR, et al. Distinct synovial tissue macrophage subsets regulate inflammation and remission in rheumatoid arthritis. Nat Med. 2020;26(8):1295–306.
Kuo D, Ding J, Cohn IS, Zhang F, Wei K, Rao DA, et al. HBEGF+ macrophages in rheumatoid arthritis induce fibroblast invasiveness. Sci Transl Med. 2019;11(491):eaau8587.
Yan M, Sun Z, Wang J, Zhao H, Yu T, Zhang Y, et al. Single-cell RNA sequencing reveals distinct chondrocyte states in femoral cartilage under weight-bearing load in rheumatoid arthritis. Front Immunol. 2023;14:1247355.
Andreev D, Liu M, Kachler K, Llerins Perez M, Kirchner P, Kölle J, et al. Regulatory eosinophils induce the resolution of experimental arthritis and appear in remission state of human rheumatoid arthritis. Ann Rheum Dis. 2021;80(4):451–68.
Zhang W, Noller K, Crane J, Wan M, Wu X, Cahan P, et al. RANK + TLR2 + myeloid subpopulation converts autoimmune to joint destruction in rheumatoid arthritis. Elife. 2023;12:e85553.
Wei K, Korsunsky I, Marshall JL, Gao A, Watts GFM, Major T, et al. Notch signalling drives synovial fibroblast identity and arthritis pathology. Nature. 2020;582(7811):259–64.
Croft AP, Campos J, Jansen K, Turner JD, Marshall J, Attar M, et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature. 2019;570(7760):246–51.
Chen N, Fan B, He Z, Yu X, Wang J. Identification of HBEGF+ fibroblasts in the remission of rheumatoid arthritis by integrating single-cell RNA sequencing datasets and bulk RNA sequencing datasets. Arthritis Res Ther. 2022;24(1):215.
Farina L, Minnone G, Alivernini S, Caiello I, MacDonald L, Soligo M, et al. Pro nerve growth factor and its receptor p75NTR activate inflammatory responses in synovial fibroblasts: a novel targetable mechanism in arthritis. Front Immunol. 2022;13:818630.
Wu J, Feng Z, Chen L, Li Y, Bian H, Geng J, et al. TNF antagonist sensitizes synovial fibroblasts to ferroptotic cell death in collagen-induced arthritis mouse models. Nat Commun. 2022;13(1):676.
Smith MH, Gao VR, Periyakoil PK, Kochen A, DiCarlo EF, Goodman SM, et al. Drivers of heterogeneity in synovial fibroblasts in rheumatoid arthritis. Nat Immunol. 2023;24(7):1200–10.
Biesemann N, Margerie D, Asbrand C, Rehberg M, Savova V, Agueusop I, et al. Additive efficacy of a bispecific anti-TNF/IL-6 nanobody compound in translational models of rheumatoid arthritis. Sci Transl Med. 2023;15(681):eabq4419.
Meng X, Chen Z, Li T, Nie Z, Han H, Zhong S, et al. Role and therapeutic potential for targeting fibroblast growth factor 10/FGFR1 in relapsed rheumatoid arthritis. Arthritis Rheumatol. 2024;76(1):32–47.
Zhou BO, Yue R, Murphy MM, Peyer JG, Morrison SJ. Leptin-receptor-expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell. 2014;15(2):154–68.
Chan CK, Seo EY, Chen JY, Lo D, McArdle A, Sinha R, et al. Identification and specification of the mouse skeletal stem cell. Cell. 2015;160(1–2):285–98.
Worthley DL, Churchill M, Compton JT, Tailor Y, Rao M, Si Y, et al. Gremlin 1 identifies a skeletal stem cell with bone, cartilage, and reticular stromal potential. Cell. 2015;160(1–2):269–84.
Debnath S, Yallowitz AR, McCormick J, Lalani S, Zhang T, Xu R, et al. Discovery of a periosteal stem cell mediating intramembranous bone formation. Nature. 2018;562(7725):133–9.
Chan C, Gulati G, Sinha R, Tompkins J, Lopez M, Carter A, et al. Identif Hum Skeletal Stem Cell Cell. 2018;175(1):43–56.e21.
Ambrosi TH, Marecic O, McArdle A, Sinha R, Gulati GS, Tong X, et al. Aged skeletal stem cells generate an inflammatory degenerative niche. Nature. 2021;597(7875):256–62.
Yin Z, Lin J, Yan R, Liu R, Liu M, Zhou B, et al. Atlas of musculoskeletal stem cells with the soft and hard tissue differentiation architecture. Adv Sci (Weinh). 2020;7(23):2000938.
Hao RC, Li ZL, Wang FY, Tang J, Li PL, Yin BF, et al. Single-cell transcriptomic analysis identifies a highly replicating Cd168+ skeletal stem/progenitor cell population in mouse long bones. J Genet Genomics. 2023;50(9):702–12.
Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505(7483):327–34.
Einhorn TA, Gerstenfeld LC. Fracture healing: mechanisms and interventions. Nat Rev Rheumatol. 2015;11(1):45–54.
Julien A, Kanagalingam A, Martínez-Sarrà E, Megret J, Luka M, Ménager M, et al. Direct contribution of skeletal muscle mesenchymal progenitors to bone repair. Nat Commun. 2021;12(1):2860.
Julien A, Perrin S, Martinez-Sarra E, Kanagalingam A, Carvalho C, Luka M, et al. Skeletal stem/progenitor cells in periosteum and skeletal muscle share a common molecular response to bone injury. J Bone Min Res. 2022;37(8):1545–61.
Serowoky MA, Kuwahara ST, Liu S, Vakhshori V, Lieberman JR, Mariani FV. A murine model of large-scale bone regeneration reveals a selective requirement for Sonic hedgehog. NPJ Regenerative Med. 2022;7(1):30.
Matsushita Y, Nagata M, Kozloff KM, Welch JD, Mizuhashi K, Tokavanich N, et al. A wnt-mediated transformation of the bone marrow stromal cell identity orchestrates skeletal regeneration. Nat Commun. 2020;11(1):332.
Novack DV, Teitelbaum SL. The osteoclast: friend or foe? Annu Rev Pathol. 2008;3:457–84.
Luo Y, Wu W, Gu J, Zhang X, Dang J, Wang J, et al. Human gingival tissue-derived MSC suppress osteoclastogenesis and bone erosion via CD39-adenosine signal pathway in autoimmune arthritis. EBioMedicine. 2019;43:620–31.
Wu W, Xiao Z, Chen Y, Deng Y, Zeng D, Liu Y, et al. CD39 produced from human GMSCs regulates the balance of osteoclasts and osteoblasts through the Wnt/β-catenin pathway in osteoporosis. Mol Ther. 2020;28(6):1518–32.
Pingping B, Yuhong Z, Weiqi L, Chunxiao W, Chunfang W, Yuanjue S, et al. Incidence and mortality of sarcomas in Shanghai, China, during 2002–2014. Front Oncol. 2019;9:662.
Guo J, Tang H, Huang P, Guo J, Shi Y, Yuan C, et al. Single-cell profiling of tumor microenvironment heterogeneity in osteosarcoma identifies a highly invasive subcluster for predicting prognosis. Front Oncol. 2022;12:732862.
Huang X, Wang L, Guo H, Zhang W, Shao Z. Single-cell transcriptomics reveals the regulative roles of cancer associated fibroblasts in tumor immune microenvironment of recurrent osteosarcoma. Theranostics. 2022;12(13):5877–87.
Broehm CJ, Garbrecht EL, Wood J, Bocklage T. Two cases of sarcoma arising in giant cell tumor of bone treated with denosumab. Case Rep Med. 2015;2015:767198.
Zhou Y, Yang D, Yang Q, Lv X, Huang W, Zhou Z, et al. Single-cell RNA landscape of intratumoral heterogeneity and immunosuppressive microenvironment in advanced osteosarcoma. Nat Commun. 2020;11(1):6322.
Feng W, He M, Jiang X, Liu H, Xie T, Qin Z, et al. Single-cell RNA sequencing reveals the migration of osteoclasts in giant cell tumor of bone. Front Oncol. 2021;11:715552.
Yin H, Price F, Rudnicki MA. Satellite cells and the muscle stem cell niche. Physiol Rev. 2013;93(1):23–67.
Giordani L, He GJ, Negroni E, Sakai H, Law JYC, Siu MM, et al. High-dimensional single-cell cartography reveals novel skeletal muscle-resident cell populations. Mol Cell. 2019;74(3):609–21.e6.
Andre AB, Rees KP, O’Connor S, Severson GW, Newbern JM, Wilson-Rawls J, et al. Single cell analysis reveals satellite cell heterogeneity for proinflammatory chemokine expression. Front Cell Dev Biol. 2023;11:1084068.
Penaloza JS, Pappas MP, Hagen HR, Xie N, Chan SSK. Single-cell RNA-seq analysis of Mesp1-induced skeletal myogenic development. Biochem Biophys Res Commun. 2019;520(2):284–90.
Yang W, Wang Y, Du Y, Li J, Jia M, Li S, et al. Chemical reprogramming of melanocytes to skeletal muscle cells. J Cachexia Sarcopenia Muscle. 2023;14(2):903–14.
Wosczyna MN, Rando TA. A muscle stem cell support group: coordinated cellular responses in muscle regeneration. Dev Cell. 2018;46(2):135–43.
De Micheli AJ, Laurilliard EJ, Heinke CL, Ravichandran H, Fraczek P, Soueid-Baumgarten S, et al. Single-cell analysis of the muscle stem cell hierarchy identifies heterotypic communication signals involved in skeletal muscle regeneration. Cell Rep. 2020;30(10):3583–95.e5.
Xi H, Langerman J, Sabri S, Chien P, Young CS, Younesi S, et al. A human skeletal muscle atlas identifies the trajectories of stem and progenitor cells across development and from human pluripotent stem cells. Cell Stem Cell. 2020;27(1):158–76.e10.
Guo R, You X, Meng K, Sha R, Wang Z, Yuan N, et al. Single-cell RNA sequencing reveals heterogeneity of Myf5-derived cells and altered myogenic fate in the absence of SRSF2. Adv Sci (Weinh). 2022;9(18):e2105775.
Scott RW, Arostegui M, Schweitzer R, Rossi FMV, Underhill TM. Hic1 defines quiescent mesenchymal progenitor subpopulations with distinct functions and fates in skeletal muscle regeneration. Cell Stem Cell. 2019;25(6):797–813.e9.
Yan J, Yang Y, Fan X, Liang G, Wang Z, Li J, et al. circRNAome profiling reveals circFgfr2 regulates myogenesis and muscle regeneration via a feedback loop. J Cachexia Sarcopenia Muscle. 2022;13(1):696–712.
Kaji DA, Montero AM, Patel R, Huang AH. Transcriptional profiling of mESC-derived tendon and fibrocartilage cell fate switch. Nat Commun. 2021;12(1):4208.
Harvey T, Flamenco S, Fan CM. A Tppp3+Pdgfra+ tendon stem cell population contributes to regeneration and reveals a shared role for PDGF signalling in regeneration and fibrosis. Nat Cell Biol. 2019;21(12):1490–503.
Fan C, Zhao Y, Chen Y, Qin T, Lin J, Han S, et al. A Cd9+Cd271+ stem/progenitor population and the SHP2 pathway contribute to neonatal-to-adult switching that regulates tendon maturation. Cell Rep. 2022;39(4):110762.
Muscat S, Nichols AEC, Gira E, Loiselle AE. CCR2 is expressed by tendon resident macrophage and T cells, while CCR2 deficiency impairs tendon healing via blunted involvement of tendon-resident and circulating monocytes/macrophages. FASEB J. 2022;36(11):e22607.
Cao ZJ, Wei L, Lu S, Yang DC, Gao G. Searching large-scale scRNA-seq databases via unbiased cell embedding with cell BLAST. Nat Commun. 2020;11(1):3458.
Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10(12):1213–8.
Ståhl PL, Salmén F, Vickovic S, Lundmark A, Navarro JF. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. 2016;353(6294):78–82.
Stephenson E, Webb S, Haniffa M. Multiomics uncovers developing immunological lineages in human. Eur J Immunol. 2021;51(4):764–72.
Wagner DE, Klein AM. Lineage tracing meets single-cell omics: opportunities and challenges. Nat Rev Genet. 2020;21(7):410–27.
Bowling S, Sritharan D, Osorio FG, Nguyen M, Cheung P, Rodriguez-Fraticelli A, et al. An engineered CRISPR-Cas9 mouse line for simultaneous readout of lineage histories and gene expression profiles in single cells. Cell. 2020;181(6):1410–22.e27.
Li L, Bowling S, McGeary SE, Yu Q, Lemke B, Alcedo K, et al. A mouse model with high clonal barcode diversity for joint lineage, transcriptomic, and epigenomic profiling in single cells. Cell. 2023;186(23):5183–99.e22.
Acknowledgements
The authors are grateful to Dan-Ni Yue for her insightful suggestions on enhancing the graphical representations.
Funding
This work was supported by the National Key Research and Development Program of China (2022YFA1103202), the National Natural Science Foundation of China (82272507, 32270887, and 32200654), the Natural Science Foundation of Chongqing (CSTB2023NSCQ-ZDJO008), the Postdoctoral Innovative Talent Support Program (BX20220397), the Independent Research Project of State Key Laboratory of Trauma and Chemical Poisoning (SFLKF202201), Project for Enhancing Innovation of Army Medical University (2023X1839), Talent Innovation Training Program at the Army Medical Center (ZXZYTSYS09) and the General Hospital of Western Theater Command Research Project (2021-XZYG-B10). This study is partially supported by the University Grants Committee, Research Grants Council of Hong Kong, China (14113723, N_CUHK472/22, C7030-18G, T13-402/17-N and AoE/M-402/20).
Author information
Authors and Affiliations
Contributions
PL and JXJ conceived and coordinated the study, contributing significantly to the manuscript drafting. PL in drafted the manuscript and designed the figures. YBG and JH played a crucial role in revising the figures and the manuscript. JZ, SH, OH, YBW, HJJ, PLY, and YYL offered assistance in manuscript drafting. SEL, JKX, LC, LMZ, and LC provided insightful suggestions for manuscript improvement. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lin, P., Gan, YB., He, J. et al. Advancing skeletal health and disease research with single-cell RNA sequencing. Military Med Res 11, 33 (2024). https://doi.org/10.1186/s40779-024-00538-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40779-024-00538-3