
Abstract
Background
This study aimed to investigate the gene expression changes associated with carcinoma-associated fibroblasts (CAFs) involving in non-small cell lung carcinoma (NSCLC).
Methods
We downloaded the GEO series GSE22862, which contained matched gene expression values for 15 CAF and normal fibroblasts samples, and series GSE27289 containing SNP genotyping for four matched NSCLC samples. The differentially expressed genes in CAF samples were identified using the limma package in R. Then we performed gene ontology (GO) and pathway enrichment analysis and protein–protein interaction (PPI) network construction using the identified DEGs. Moreover, aberrant cell fraction, ploidy, allele-specific copy number, and loss of heterozygosity (LOH) within CAF cells were analyzed using the allele-specific copy number analysis.
Results
We obtained 545 differentially expressed genes between CAF and normal fibroblasts samples. The up-regulated genes are mainly involved in GO terms such as positive regulation of cell migration and extracellular region, while the down-regulated genes participate in the lung development and extracellular region. Multiple genes including bone morphogenetic protein 4 (BMP4) and transforming growth factor, beta 3 (TGFB3) are involved in the TGF-β signaling pathway. Genes including BMP4, TGFBI and matrix Gla protein (MGP) were hub genes. Moreover, no LOH event for BMP4 and MGP was found, that for sphingosine kinase 1 (SPHK1) was 70%, and for TGFBI was 40%.
Conclusion
Our data suggested that BMP4, MGP, TGFBI, and SPHK1 may be important in CAFs-associated NSCLC, and the abnormal expression and high LOH frequency of them may be used as the diagnosis targets of CAFs in NSCLC.
Similar content being viewed by others

Background
Lung cancer is one of the most common leading cause of cancer deaths worldwide [1]. Statistics show that the non-small cell lung carcinoma (NSCLC) accounts for about 85% of all lung cancers in the world [2]. Treatment methods such as surgery, drug therapy and chemotherapy have played certain roles in curing NSCLC [2]. However, in [3] and [4], the authors suggest that 5-year survival rate is poor due to the difficulties in the early diagnose of NSCLC and the easy invasion and metastasis of NSCLC cells. Therefore, exploring the mechanism in NSCLC metastasis and invasion will be of great significance to provide basis for NSCLC diagnosis and treatment.
In [5], authors suggest that carcinoma-associated fibroblasts (CAFs) are the most important component of developing cancers. Recent studies implied that CAFs played important roles in cancers biology occurring in epithelia, such as neoplastic progression, tumor growth, invasion and metastasis [6,7,8]. CAFs constitute a major portion of the reactive tumor stroma and are related to the cell invasion and metastasis in NSCLC during malignancy [9]. Also, Neta et al. [10] referred that CAFs activities promoted the macrophage recruitment, neovascularization, and tumor growth in incipient neoplasia to orchestrate tumor inflammation via the NF-kB signaling pathway. Furthermore, an increasing number of evidences show that mutations of genes for CAFs are crucial for cancer metastasis and growth. For examples, some changes in DNA copy number of cluster CAFs may contribute to the metastasis for breast cancer cells [11]. In spite of many researches devoted to the exploration of the role of CAFs in NSCLC, the mechanism of CAFs in NSCLC still remains not fully understood.
Using the GEO (Gene Expression Omnibus) series GSE22862, Horie et al. [12] proved that RNAi-mediated targeting of transforming growth factor, beta 1 (TGFB1) ligands were beneficial for lung cancer treatment through its action on cancer and stromal cells. In this study, we screened the differentially expressed genes (DEGs) in NSCLC samples compared with the normal samples using the same GEO series GSE22862 [13]. Bioinformatics methods were used to analyze the functions and pathways of the DEGs, as well as the allele-specific copy number (ASCN) of them to predict hub genes which were related with the CAFs in NSCLC. This study aimed to explore the underlying genes associated with pathomechanism of CAFs in NSCLC, which may help to search for diagnosis and treatment targets for this disease.
Methods
Microarray data
We downloaded the GEO series GSE22874 [13] from the GEO database in NCBI (http://www.ncbi.nlm.nih.gov/geo/). This series contains four subseries and two of them (GSE22862 and GSE27289) were analyzed in this study. The single nucleotide polymorphism (SNP) series GSE27289 is generated from four paired primary NSCLC CAF and normal fibroblasts (NF) samples based on the platform of GPL13135 HumanOmniExpress BeadChip. GSE22862 is a CAF series, which originates from matched gene expression values from 15 CAF and NF samples of resected NSCLC tissues based on the platform of GPL5175 Affymetrix Human Exon 1.0 ST Array (Affymetrix Inc., Santa Clara, California, USA). At present, we chose four matched samples (4 CAF samples and 4 NF samples) of two stages I squamous cell carcinomas and 2 stage II adenocarcinoma from the 30 samples of GSE28682.
Microarray data preprocessing and conversion
The CEL file data of GSE22862 download from the GEO database have been normalized using the Robust Multi-array Analysis method [14] in affy (http://www.bioconductor.org/packages/release/bioc/html/affy.html) package in Bioconductor. When multiple probes correspond to the same gene, the mean expression value was calculated and considered as the expression value of this gene. Meanwhile, the CEL file data of GSE27289 that have been preprocessed using the GenomeStudio [15], were extracted for the SNP symbols, chromosome number, chromosome location, B allele frequency and Log R values both in CAF samples and in NF samples.
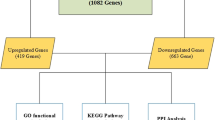
Screening and functional enrichment analysis of DEGs
We screened the DEGs between CAF and NF samples in GSE22862 series using the limma package in R (http://www.bioconductor.org). The p value <0.05 and |log2 (fold change)| ≥0.58 (fold change ≥1.5 or ≤0.67) were chosen as the thresholds. In the present study, we did not performed multiple test correction for the p values, because the corrected p values were too low to select enough significant genes.
In addition, we conducted gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways enrichment analyses for the DEGs using the Database for Annotation Visualization and Integrated Discovery (DAVID, http://david.abcc.ncifcrf.gov/) [16] online tool. p value <0.05 was chosen as threshold.
Protein–protein interaction (PPI) network construction of DEGs
PPI analysis can provide new insights into protein function, besides, it may help to uncover the generic organization principles of functional cellular networks [17], therefore, we would construct PPI network to further analyze the DEGs. Search Tool for the Retrieval of Interacting Genes/Proteins (STRING, http://string.embl.de/) [18] that provided functional associations between proteins was used to predict the interaction pairs of the selected DEGs. In this study, we only mapped the DEGs into STRING database to predict the PPI pairs because we intended to investigate the interactions between DEGs. Cytoscape (http://www.cytoscape.org/) [19] that was widely used to integrate biomolecular interaction networks into models was used to construct the PPI network of the DEGs. Most of previous obtained biological networks were found to obey the scale-free attribution [20]. Thus, we analyzed the connectivity degree of nodes in the PPI network by topological analysis to obtain the important nodes with higher degrees (hub proteins) [21]. Genome-wide studies have shown that deletion of a hub protein is more likely to be lethal than deletion of a non-hub protein, thus, we think that the hub nodes may play important roles in the CAF of NSCLC.
ASCN analysis
ASCN analysis of tumor [22, 23] is a method that can be used to analyze the aberrant cell fraction, ploidy, ASCN, and loss of heterozygosity (LOH) of tumor cells. The ASCN for one gene was calculated based on the Log R and B allele frequency values of allele B. In this study, we used ASCN method to determine the values of the aberrant cell fraction, ploidy, ASCN and LOH of the selected genes. Twenty-two autosomes plus two gender associated chromosomes were chosen as parameters of the number of the chromosomes.
Candidate gene identification
On account of GO and KEGG pathway enrichment results, as well as PPI network of the DEGs, we identified the potential critical genes combining with the results of ASCN analysis. Additionally, based on the databases of tumor suppressor (TS) gene [9] and tumor-associated gene (TAG) [10], we identified the DEGs that function as transcription factors, tumor suppressors or oncogenes. Finally, the ASCN values of these identified genes were analyzed.
Results
Identified DEGs
With p value <0.05 and |log2 (fold change)| ≥0.58, we identified 545 DEGs including 66 up-regulated and 479 down-regulated DEGs in CAF samples compared with NF samples in GSE22862. The number of down-regulated DEGs was far more than that of up-regulated DEGs, suggesting the important roles of down-regulated genes in NSCLC.
Functional enrichment analysis of DEGs
The GO functions and pathways of up- and down-regulated DEGs in CAF samples were shown in Tables 1, 2, respectively. Up-regulated genes are significantly involved in GO terms such as cell surface receptor linked signal transduction, G-protein couple receptor protein, positive regulation of cell migration and extracellular region, while down-regulated genes are associated with GO terms including lung development, respiratory tube development and extracellular region (Table 1).
In addition, only pathway of Olfactory transduction is enriched by up-regulated genes, such as olfactory receptor, family 6, subfamily B, member 1 (OR6B1), olfactory receptor, family 5, subfamily L, member 2 (OR5L2) and olfactory receptor, family 10, subfamily H, member 1 (OR10H1). The down-regulated genes, such as bone morphogenetic protein 4 (BMP4), TGFB3 and SMAD family member 3 (SMAD3), are mainly involved in TGF-β signaling pathway (Table 2).
PPI network construction
The PPI network of DEGs was constructed and shown in Fig. 1. The PPI network contained 266 nodes and 409 interaction pairs. Among the top ten high degree genes, only two were up-regulated, including toll-like receptor 4 (TLR4, degree = 12) and chemokine (C-X-C Motif) ligand 10 (CXCL10, degree = 11). The other eight down-regulated genes including transforming growth factor, beta-induced (TGFBI, degree = 19), neurotensin (NTS, degree = 15), matrix Gla Protein (MGP, degree = 13), guanine nucleotide binding protein (G Protein), gamma 2 (GNG2, degree = 13), collagen, type III, alpha 1 (COL3A1, degree = 13), BMP4 (degree = 12), peroxisome proliferator-activated receptor (PPARG, degree = 12), and collagen, type XIV, alpha 1 (COL14A1, degree = 11).

Protein–protein interaction network of the selected differentially expressed genes (DEGs). Red circle stands for the up-regulated genes while green circle stands for the down-regulated genes. Gray circle stands for the non-differentially expressed genes. Edge stands for the interaction between two genes
Analysis of copy number of alleles
The aberrant cell fraction and ploidy of genes of tumor cells in CAF samples were respectively 77% and 2.002 based on the ASCNT analysis. In addition, based on the functional enrichment analysis and PPI network, we obtained 16 critical genes. ASCN results of the 16 critical genes were shown in Table 3. The copy number of alleles of most genes were abnormal except for MGP, BMP4, PPARG and TLR4, moreover, they were all positive for LOH. For instance, the LOH proportion of TGFB3 and sphingosine kinase 1 (SPHK1) was 70%, while that of MGP was 0% (Table 3).
Identified candidate genes
The result of function annotation for DEGs in CAF samples was shown in Table 4. The results displayed that six up-regulated genes and ten down-regulated genes function as transcription factors. In addition, SPHK1 and pre-B-cell leukemia homeobox 1 (PBX1), etc. were oncogenes, while runt-related transcription factor 3 (RUNX3) and TGFBI, etc. were tumor suppressor genes (Table 4).
The result of ASCN analysis of genes mentioned above was shown in Table 5. As shown in Table 5, LOH of latexin (LXN), RAS-like, family 11, member A (RASL11A) and potassium channel regulator (KCNRG) were all 100%, LOH of transcription factor ap-2 gamma (TFAP2C) and SPHK1 was 70%, of tumor protein P53 inducible nuclear protein 1 (TP53INP1) was 60%, and of homeobox D12 (HOXD12), ERBB receptor feedback inhibitor 1 (ERRFI1), ribosomal protein S6 kinase, 90 kDa, polypeptide 6 (RPS6KA6), DnaJ (Hsp40) homolog, subfamily B, member 4 (DNAJB4) and integrin, alpha 7 (ITGA7) was 50%. Moreover, the copy numbers of allele for these genes were not the same (Table 5).
Discussion
In this study, we screened a total of 66 up-regulated and 479 down-regulated DEGs by comparing gene expression between CAFs tissues and NF tissues in NSCLC. DEGs including BMP4, TGFB3 and MGP involving in the TGF-β signaling pathway were found to be hub genes in the PPI network. Moreover, no LOH was found in both BMP4 and MGP. Besides, LOH of the tumor oncogene SPHK1 was 70%, and of tumor suppressor gene TGFBI was 40%.
BMP4 is a member of the bone morphogenetic protein family which belongs to the transforming growth factor (TGF-β) superfamily [24], while no LOH events were found about it. In addition, it was found that BMP4 was a hub gene in the PPI network and involved in lung development and the TGF-β signaling pathway. Meiou et al. [25] reported that the TGF-β factor signaling which was regulated by the transcriptional co-activator p/CAF (a histone acetyltransferase), played key roles in breast cancer cell migration and invasion. In previous studies, BMP4 was found play important roles in developmental and many cellular processes including invasion and migration of various cancer cells [26, 27]. Since BMP2 is closely associated with NSCLC metastasis [28], BMP4 may be a contributor to affect the behavior and function of CAFs in NSCLC via TGF-β signaling pathway. Meanwhile, TGFB3 as another member of the TGF-β family is also enriched in lung development and TGF-β signaling pathway. It was detected with a high frequency of LOH (70%). Some studies indicated the abnormal expression of TGFB3 in NSCLC [29, 30]. Therefore, abnormal expression of BMP4 and TGFB3, as well the high LOH frequency of TGFB3 may be used as biological indicators for malignant CAFs in NSCLC.
Our data also showed that tumor suppressor TGFBI was a hub protein in the PPI network and LOH value of it was 40%. TGFBI has been reported to suppress tumor cell growth in NSCLC [31] and other types of human lung cancers [32]. Therefore, TGFBI may be a tumor suppressor for NSCLC. Besides, Muraoka et al. [33] proved that blocking TGFBI expression enhanced inhibition of tumor cell migration and metastasis. Also, Levy et al. [34] proved that LOH of TGFBI located on chromosome 19q13.1 contributed to the metastasis of breast cancer cells. Thus, LOH may lead to the down-regulation of TGFBI in this study. On the other hand, Fong et al. [35] said that TGFBI promoted the migration of lung cancer cells. CAFs are important for breast cancer cell migration and metastasis [36]. Based on the previous evidences, we speculated that TGFBI may be a tumor suppressor for NSCLC and may be responsible for the CAFs migration in NSCLC.
SPHK1 catalyzes the phosphorylation of sphingosine to form sphingosine-1-phosphate (S1P), a lipid mediator with both intra- and extracellular functions [37]. Presneau and his colleagues proved that the LOH of SPHK1 assigned to chromosome region at 17q25.1-q25.2, suggesting its key role in regulating early events during the development of sensory ganglia in ovarian cancer [38]. Thus, the higher LOH value may be related to the abnormal expression of SPHK1 in NSCLC samples. Therefore, the positive result of SPHK1 at LOH analysis was consistent with the up-regulation of it. In addition, GO analysis showed that SPHK1 enriched in multiple cancer-related processes including positive regulation of cell migration. Chen et al. [39] reported that the tumor oncogene SPHK1 which was up-regulated was responsible for tumor cell migration in hepatocellular carcinoma. Moreover, SPHK1 was found to enhance the NSCLC cell apoptosis via activating PI3K/Akt pathway [40]. Besides, it can induce breast cancer cell migration [41]. Therefore, we speculated that over-expressed SPHK1 may be associated with CAFs migration in NSCLC.
Interestingly, some of our results were in accordance with the original publication by Navab et al. [13]. In their study, microarray gene-expression analysis of the 15 matched CAF and NF cell lines identifies 46 differentially expressed genes which are significantly enriched for extracellular proteins regulated by the TGF-β signaling pathway. Our study also found that BMP4 and TGFB3 are involved in TGF-β signaling pathway, suggesting the important role of this pathway in CAF-associated NSCLC. Additionally, their study revealed prominent involvement of the focal adhesion and MAPK signaling pathways, which was not identified in our study. This difference may be due to different sample size and different screening threshold for DEGs.
In conclusion, genes such as BMP4, TGFB3, TGFBI, and SPHK1 may play important roles in CAFs of NSCLC. BMP4 and TGFB3 may be contributor to make effects on CAFs in NSCLC via TGF-β signaling pathway. SPHK1 promoted while TGFBI inhibited the NSCLC progression, and both of them may be associated with CAFs migration in NSCLC. In addition, high frequency of TGFB3, TGFBI and SPHK1 may be useful for distinguishing the normal fibroblasts from malignant CAFs in NSCLC. However, further experimental studies are still needed to confirm our results.
Abbreviations
- ASCN:
-
allele-specific copy number
- BMP4:
-
bone morphogenetic protein 4
- CAFs:
-
carcinoma-associated fibroblasts
- DEGs:
-
differentially expressed genes
- GO:
-
gene ontology
- LOH:
-
loss of heterozygosity
- MGP:
-
matrix Gla protein
- NSCLC:
-
non-small cell lung carcinoma
- PPI:
-
protein–protein interaction
- RUNX3:
-
runt-related transcription factor 3
- SPHK1:
-
sphingosine kinase 1
- TGF-β:
-
transforming growth factor
- TGFB3:
-
transforming growth factor, beta 3
- TP53INP1:
-
tumor protein P53 inducible nuclear protein 1
- TS:
-
tumor suppressor
References
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30.
Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, Palmero R, Garcia-Gomez R, Pallares C, Sanchez JM. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13(3):239–46.
Temel JS, Greer JA, Muzikansky A, Gallagher ER, Admane S, Jackson VA, Dahlin CM, Blinderman CD, Jacobsen J, Pirl WF. Early palliative care for patients with metastatic non-small-cell lung cancer. N Engl J Med. 2010;363(8):733–42.
Breindel JL, Haskins JW, Cowell EP, Zhao M, Nguyen DX, Stern DF. EGF receptor activates MET through MAPK to enhance non-small cell lung carcinoma invasion and brain metastasis. Cancer Res. 2013;73(16):5053–65.
Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007;67(21):10123–8.
Mishra PJ, Mishra PJ, Humeniuk R, Medina DJ, Alexe G, Mesirov JP, Ganesan S, Glod JW, Banerjee D. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008;68(11):4331–9.
Liu T, Lin B, Qin J. Carcinoma-associated fibroblasts promoted tumor spheroid invasion on a microfluidic 3D co-culture device. Lab Chip. 2010;10(13):1671–7.
Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu C-C, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer cell. 2014;25(6):719–34.
Bremnes RM, Dønnem T, Al-Saad S, Al-Shibli K, Andersen S, Sirera R, Camps C, Marinez I, Busund L-T. The role of tumor stroma in cancer progression and prognosis: emphasis on carcinoma-associated fibroblasts and non-small cell lung cancer. J Thorac Oncol. 2011;6(1):209–17.
Erez N, Truitt M, Olson P, Hanahan D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-κB-dependent manner. Cancer Cell. 2010;17(2):135–47.
Hosein AN, Wu M, Arcand SL, Lavallée S, Hébert J, Tonin PN, Basik M. Breast carcinoma-associated fibroblasts rarely contain p53 mutations or chromosomal aberrations. Cancer Res. 2010;70(14):5770–7.
Horie M, Saito A, Noguchi S, Yamaguchi Y, Ohshima M, Morishita Y, Suzuki HI, Kohyama T, Nagase T. Differential knockdown of TGF-β ligands in a three-dimensional co-culture tumor-stromal interaction model of lung cancer. BMC Cancer. 2014;14(1):580.
Navab R, Strumpf D, Bandarchi B, Zhu C-Q, Pintilie M, Ramnarine VR, Ibrahimov E, Radulovich N, Leung L, Barczyk M. Prognostic gene-expression signature of carcinoma-associated fibroblasts in non-small cell lung cancer. Proc Natl Acad Sci. 2011;108(17):7160–5.
Wu Z, Irizarry RA. Preprocessing of oligonucleotide array data. Nat Biotechnol. 2004;22(6):656–8.
Gentilini D, Mari D, Castaldi D, Remondini D, Ogliari G, Ostan R, Bucci L, Sirchia SM, Tabano S, Cavagnini F. Role of epigenetics in human aging and longevity: genome-wide DNA methylation profile in centenarians and centenarians’ offspring. Age. 2013;35(5):1961–73.
Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4(5):P3.
Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, Goehler H, Stroedicke M, Zenkner M, Schoenherr A, Koeppen S. A human protein–protein interaction network: a resource for annotating the proteome. Cell. 2005;122(6):957–68.
Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C. STRING v9. 1: protein–protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41(D1):D808–15.
Smoot ME, Ono K, Ruscheinski J, Wang P-L, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27(3):431–2.
Lim J, Hao T, Shaw C, Patel AJ, Szabó G, Rual J-F, Fisk CJ, Li N, Smolyar A, Hill DE. A protein–protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell. 2006;125(4):801–14.
He X, Zhang J. Why do hubs tend to be essential in protein networks? PLoS Genet. 2006;2(6):e88.
Van Loo P, Nordgard SH, Lingjærde OC, Russnes HG, Rye IH, Sun W, Weigman VJ, Marynen P, Zetterberg A, Naume B. Allele-specific copy number analysis of tumors. Proc Natl Acad Sci. 2010;107(39):16910–5.
Nilsen G, Liestøl K, Van Loo P, Vollan HKM, Eide MB, Rueda OM, Chin S-F, Russell R, Baumbusch LO, Caldas C. Copynumber: efficient algorithms for single-and multi-track copy number segmentation. BMC Genom. 2012;13(1):591.
Knight PG, Glister C. TGF-β superfamily members and ovarian follicle development. Reproduction. 2006;132(2):191–206.
Dai M, Al-Odaini AA, Arakelian A, Rabbani SA, Ali S, Lebrun J-J. A novel function for p21Cip1 and acetyltransferase p/CAF as critical transcriptional regulators of TGFb-mediated breast cancer cell migration and invasion. Breast Cancer Res. 2012;14(5):1–22.
Guo D, Huang J, Gong J. Bone morphogenetic protein 4 (BMP4) is required for migration and invasion of breast cancer. Mol Cell Biochem. 2012;363(1–2):179–90.
Ma W, Ma J, Xu J, Qiao C, Branscum A, Cardenas A, Baron AT, Schwartz P, Maihle NJ, Huang Y. Lin28 regulates BMP4 and functions with Oct4 to affect ovarian tumor microenvironment. Cell Cycle. 2013;12(1):88–97.
Bieniasz M, Oszajca K, Eusebio M, Kordiak J, Bartkowiak J, Szemraj J. The positive correlation between gene expression of the two angiogenic factors: VEGF and BMP-2 in lung cancer patients. Lung Cancer. 2009;66(3):319–26.
Jakowlew SB, Mathias A, Chung P, Moody TW. Expression of transforming growth factor beta ligand and receptor messenger RNAs in lung cancer cell lines. Cell Growth Differ. 1995;6(4):465–76.
Jakowlew SB, Zakowicz H, Moody TW. Retinoic acid down-regulates VPAC 1 receptors and TGF-β3 but up-regulates TGF-β2 in lung cancer cells. Peptides. 2000;21(12):1831–7.
Asselin-Paturel C, Echchakir H, Carayol G, Gay F, Opolon P, Grunenwald D, Chouaib S, Mami-Chouaib F. Quantitative analysis of Th1, Th2 and TGF-β1 cytokine expression in tumor, TIL and PBL of non-small cell lung cancer patients. Int J Cancer. 1998;77(1):7–12.
Sumitomo K, Kurisaki A, Yamakawa N, Tsuchida K, Shimizu E, Sone S, Sugino H. Expression of a TGF-β1 inducible gene, TSC-36, causes growth inhibition in human lung cancer cell lines. Cancer Lett. 2000;155(1):37–46.
Muraoka RS, Dumont N, Ritter CA, Dugger TC, Brantley DM, Chen J, Easterly E, Roebuck LR, Ryan S, Gotwals PJ. Blockade of TGF-β inhibits mammary tumor cell viability, migration, and metastases. J Clin Investig. 2002;109(12):1551–9.
Levy L, Hill CS. Alterations in components of the TGF-β superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 2006;17(1):41–58.
Fong Y-C, Hsu S-F, Wu C-L, Li T-M, Kao S-T, Tsai F-J, Chen W-C, Liu S-C, Wu C-M, Tang C-H. Transforming growth factor-β1 increases cell migration and β1 integrin up-regulation in human lung cancer cells. Lung Cancer. 2009;64(1):13–21.
Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6(5):392–401.
Pappu R, Schwab SR, Cornelissen I, Pereira JP, Regard JB, Xu Y, Camerer E, Zheng Y-W, Huang Y, Cyster JG. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science. 2007;316(5822):295–8.
Presneau N, Dewar K, Forgetta V, Provencher D, Mes-Masson AM, Tonin PN. Loss of heterozygosity and transcriptome analyses of a 1.2 Mb candidate ovarian cancer tumor suppressor locus region at 17q25. 1‐q25. 2. Mol Carcinog. 2005;43(3):141–54.
Chen J-S, Hung W-S, Chan H-H, Tsai S-J, Sun HS. In silico identification of oncogenic potential of fyn-related kinase in hepatocellular carcinoma. Bioinformatics. 2013;29(4):420–7.
Song L, Xiong H, Li J, Liao W, Wang L, Wu J, Li M. Sphingosine kinase-1 enhances resistance to apoptosis through activation of PI3K/Akt/NF-κB pathway in human non-small cell lung cancer. Clin Cancer Res. 2011;17(7):1839–49.
Sarkar S, Maceyka M, Hait NC, Paugh SW, Sankala H, Milstien S, Spiegel S. Sphingosine kinase 1 is required for migration, proliferation and survival of MCF-7 human breast cancer cells. FEBS Lett. 2005;579(24):5313–7.
Authors’ contributions
XW participated in the design of this study. LR performed the statistical analysis. YY carried out the study, together with QM, and collected important background information. QM drafted the manuscript. XW conceived of this study, and participated in the design and helped to draft the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The microarray data GSE22874 were downloaded from the GEO (Gene Expression Omnibus) database in NCBI (http://www.ncbi.nlm.nih.gov/geo/).
Funding
This study was supported by Hubei Provincial Natural Science Foundation of China (No. 2014CFB366).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wu, X., Ruan, L., Yang, Y. et al. Analysis of gene expression changes associated with human carcinoma-associated fibroblasts in non-small cell lung carcinoma. Biol Res 50, 6 (2017). https://doi.org/10.1186/s40659-017-0108-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40659-017-0108-9