
Abstract
Throughout the Earth’s history, mass transport involved fluids. In order to address the circumstances under which Zr4+ may have been transported in this manner, its solubility behavior in aqueous fluid with and without NaOH and SiO2 in equilibrium with crystalline ZrO2 was determined from 550 to 950 °C and 60 to 1200 MPa. The measurements were carried out in situ while the samples were at the temperatures and pressures of interest. In ZrO2-H2O and ZrO2-SiO2-H2O fluids, the Zr4+ concentration ranges from ≤10 to ~70 ppm with increasing temperature and pressure. Addition of SiO2 to the ZrO2-H2O system does not affect these values appreciably. In these two environments, Zr4+ forms simple oxide complexes in the H2O fluid with ∆H ~ 40 kJ/mol for the solution equilibrium, ZrO2(solid) = ZrO2(fluid). The Zr4+ concentration in aqueous fluid increases about an order of magnitude upon addition of 1 M NaOH, which reflects the formation of zirconate complexes. The principal solution mechanism is ZrO2 + 4NaOH = Na4ZrO4 + 2H2O with ∆H ~ 200 kJ/mol. Addition of both SiO2 and NaOH to ZrO2-H2O enhances the Zr4+ by an additional factor of about 5 with the formation of partially protonated alkali zircon silicate complexes in the fluid. The principal solution mechanism is 2ZrO2 + 2NaOH + 2SiO2 = Na2Zr2Si2O9 + H2O with ∆H ~ 40 kJ/mol. These results, in combination with other published experimental data, imply that fluid released during high-temperature/high-pressure dehydration of hydrous mineral assemblages in the Earth’s interior under some circumstances may carry significant concentrations of Zr and probably other high field strength elements (HFSEs). This suggestion is consistent with the occurrence of Zr-rich veins in high-grade metamorphic eclogite and granulite terranes. Moreover, aqueous fluids transported from dehydrating oceanic crust into overlying mantle source rocks of partial melting also may carry high-abundance HFSE of fluids released from dehydrating slabs and transported to the source rock of partial melting in the overlying mantle wedge. These processes may have been operational in the Earth’s history within which subduction resembling that observed today was operational.
Similar content being viewed by others

Background
Mass transfer in the deep crustal and upper mantle involves fluids, supercritical fluids, and melts. In the young Earth’s mantle and crust, stable isotope evidence is consistent with liquid water interacting with zircon-bearing rocks (e.g., Compston and Pidgeon 1986, Mojzsis et al. 2001, Valley et al. 2005). Early mass transfer processes were not, however, necessarily similar to more recent activities (Kröner and Layer 1992, Hamilton 2007). For example, subduction zone processes, an area of major mass transfer in more recent terrestrial environments, may not have begun until some 1–2 billion years later (Hamilton, 2007). From this time in the Earth’s history until the present day, fluid was transferred from dehydrating hydrous minerals in subduction zone materials to the overlying mantle wedge where partial melting may occur (e.g., Bebout 2007). These magmatic liquids would carry element signatures of the source of those fluids (Gill 1981, Wyllie and Sekine 1982, Tatsumi and Eggins 1995, Hermann et al. 2006).
It is often found, however, that the solubility of high field strength elements (HFSEs) such as Ti, Hf, Zr, Nb, and Ta in pure H2O fluids is in the ppm range (Brenan et al. 1994, Audetat and Keppler 2005, Antignano and Manning 2008, Newton et al. 2010, Bernini et al. 2013), which would suggest that H2O alone is not an efficient HFSE transport medium at least in the temperature/pressure range of the crust and upper mantle. There is, however, evidence from veins in high-grade metamorphic terranes that Zr and other HFSEs can be significantly mobile (Giere 1986, Philippot and Selverstone 1991, Möller et al. 2002). The latter observations have led to suggestions that complexing of HFSEs with major-element solutes in aqueous fluids would greatly enhance the HFSE solubility. Scattered solubility data of some HFSEs in water-rich multi-component fluids at high temperature and pressure are consistent with such ideas (Ayers and Watson 1993, Antignano and Manning 2008, Mysen 2012, Ayers et al. 2012, Louvel et al. 2013). Effects of major element composition on HFSE solubility in silicate melts have also been reported (Watson 1976, 1979, Ellison and Hess 1986, Linnen and Keppler 2002).
The majority of experimental studies have focused on zircon (ZrSiO4) solubility in fluids and typically on its solubility in pure H2O or in aqueous solution as a function of additional solutes (SiO2, NaOH, Na-silicates, etc.). That approach limits the interpretation of the solution mechanism of Zr4+ in fluids because the presence of SiO2 in those experiments restricts identification of effects of individual components on Zr4+ solution mechanisms. Moreover, most, although not all, characterizations of experimental charges were carried out on fluids after returning the sample to ambient temperature and pressure. This process often leads to compositional and structural changes of the fluid because solubility in aqueous fluid typically is temperature- and pressure-dependent (Pascal and Anderson 1989, Manning 1994) and, thus, changes along the pressure-temperature path of the quenching path.
In the present study, experiments were carried out in order to address some of the unanswered questions regarding the solubility and solution mechanism(s) of Zr4+ in aqueous solutions. Experimental complexities resulting from compositional and structural changes during quenching of the aqueous fluid to ambient conditions after an experiment were avoided by examination of the Zr-saturated fluids in situ while these were at the temperatures and pressures of interest. The effects of specific solutes on Zr4+ solubility and solution mechanisms in aqueous solution were addressed.
Methods
Aqueous fluids (double-distilled, deionized H2O and 1 M NaOH aqueous solutions) together with crystalline spectroscopically pure ZrO2 (Alfa Aesar™) and SiO2 (Alfa Aesar™) were used as starting materials. The grain size of the oxides was less than 5 μm. Crystalline Na2ZrO3 was synthesized from spectroscopically pure Na2CO3 + SiO2 mixtures at ambient pressure by using the method of multiple heat cycles (to ~1000 °C at ambient pressure) described by Ampian (1968). Results from X-ray diffractometry indicated the complete reaction of Na2CO3 + SiO2 to crystalline Na2ZrO3.
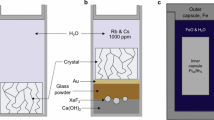
In situ, high-temperature/high-pressure experiments were carried out in externally heated, hydrothermal diamond anvil cells (HDAC) (Bassett et al. 1996). Iridium was used as gasket material in order to avoid chemical reaction between the gasket and aqueous solutions during the experiments. Gasket thickness was 125 μm with a 500 μm diameter sample hole. Temperatures were measured with two K-type thermocouples touching the upper and lower diamonds at a distance of about 1 mm from the sample. The temperature variations within the cell, as indicated by NaCl melting at ambient pressure, are 2–3 °C.
In the hydrothermal diamond cell experiments, pressure was generated by increasing the temperature of the aqueous solution + solid sample contained in the sample hole of the Ir gasket. Pressure is, therefore, a temperature-dependent variable. This temperature-dependent pressure is governed by the PVT properties of the fluid (e.g., Bassett et al. 1996). The hydrothermal diamond cell experimental protocol does not, therefore, allow for independent control of pressure. Pressure was determined with the one-phonon shift in the Raman spectra by using a 10–20 μm chip of synthetic carbon-13 diamond located within the sample region itself (Schiferl et al. 1997, Mysen and Yamashita 2010). Synthetic carbon-13 diamond was used for this purpose to avoid interference of the Raman signal from the natural diamond anvils in the diamond anvil cell. The frequency difference between the one-phonon Raman shifts from carbon-12 and carbon-13 is about 80 cm−1 (Hanfland et al. 1985).
A confocal JASCO™ Model NRS-3100 microRaman spectrometer with the 490 nm line of a Coherent MX-Series solid-state laser operating at ~40 mW at the sample for Raman excitation was used for all measurements. The Raman spectrometer is equipped with a single monochromator, holographic notch filter, and holographic gratings. For pressure calibration with a 13C diamond, gratings with 2400 grooves/mm density were used in order to optimize frequency accuracy of the spectrometer. Because of the relatively small pressure-induced shift of the Raman signal from diamond (~1.8 cm−1/GPa at ambient temperature; Hanfland et al. 1985), additional improvement in spectrometer accuracy was accomplished by using Ne emission lines as an internal standard. A ±0.1 cm−1 uncertainty thus attained translates to a ±40 MPa precision of pressure measurements. The overall pressure uncertainty, which also includes the uncertainty in the calibration of the one-phonon Raman shift of the carbon-13 diamond from Mysen and Yamashita (2010), is ±110 MPa. This uncertainty (and precision) was the same in all experiments.
Raman spectra of the samples (fluids and crystalline materials) were recorded with 1200 grooves/mm grating density, which in the frequency range of interest in the present study yields a spectral range of ~1900 cm−1 per CCD window (decreasing slightly with distance from the excitation line) and a frequency uncertainty of ~±3 cm−1. This uncertainty is not an issue given the large width of the Raman bands of fluids and crystals at the high temperatures of the experiments (full width at half height (FWHH) ≥ 30 cm−1).
Experiments were conducted in several temperature/pressure series. The proportion of fluid and solid phases in the various experimental series differs. The pressure-temperature trajectories, therefore, also differ between individual experimental series.
In each experimental series, the sample was initially brought to the highest planned temperature and pressure, left there for about an hour followed by recording the spectra of fluid and coexisting solid materials. The spectra of carbon-13 diamond for pressure determination also were recorded at each temperature to determine the pressure. The temperature was then lowered at 1 °C/s to the next temperature/pressure condition where the same spectroscopic acquisition protocol was used. A 60-minute dwell time at each temperature and pressure prior to analysis is considered sufficient to reach equilibrium given that similar experimental duration has been shown sufficient to establish isotopic equilibrium between fluid and condensed phases at similar temperatures (Horita 1988, Mysen 2015). Moreover, Wilke et al. (2012), using a similar experimental protocol, found a constant Zr concentration in fluid coexisting with crystalline ZrO2 after a 15–30-minute experimental duration.
Results
In the HDAC experiments, there is a general increase in pressure with increasing temperature (Table 1; see also Fig. 1). The “average pressure” shown in the subsequent figures is that corresponding to the intersection of the temperature with a single straight line fitted to all the data points (Fig. 1; see Tables 2 and 3 for regression parameters). The exact pressure for each data point can be extracted from the data in Table 1.

Raman spectroscopy
Raman spectra were collected in the ~300–2000 cm−1 and ~3000–4100 cm−1 frequency ranges. The higher frequency range comprises signals from fundamental OH vibrations. In the low-frequency range, Raman signals from vibrations in Zr-O and Si-O bonding environments can be detected.
The spectra in the high-frequency range characteristically show a sharp maximum near 3600 cm−1 with an asymmetry toward lower frequency, in particular at the lower temperatures (Fig. 2). For all compositions, the FWHH of this peak decreases with increasing temperature and pressure (Fig. 3). These features are qualitatively similar to the changes in the same frequency region of the Raman spectra of other aqueous fluids systems (Walrafen et al. 1986, 1999, Frantz et al. 1993).

Examples of Raman spectra of high-frequency intensity envelope. a Spectra at approximately the same temperature conditions for each of the compositions studied. b Temperature/pressure evolution example from the ZrO2-H2O system. All spectra were normalized to the total integrated area of the 3600 cm−1 envelope. Note that individual spectra are offset for clarity

The FWHH vs. temperature relations in the spectra of fluids in the ZrO2-H2O and ZrO2-SiO2-H2O systems are identical within the error of the measurements (Fig. 3). The FWHH is more sensitive to temperature with NaOH added to the H2O fluid (Tables 2 and 3). Addition of SiO2 (ZrO2-SiO2-NaOH-H2O) results in FWHH that may be slightly narrower than without SiO2 and up to >40 cm−1 narrower than in fluid spectra from the SiO2-free ZrO2-NaOH-H2O system (Fig. 3).
The vibrations in this high-frequency range (Fig. 2) are assigned to fundamental O-H stretching possibly in several locally different environments (Scholze 1960, Mysen and Virgo 1986, Walrafen et al. 1986, Le Losq et al. 2011). Their frequencies reflect which cation or cations form bonds with OH groups. Here, these cations could be H+, Na+, Si4+, and Zr4+. In addition, hydrogen bonding may play a role, in particular at temperatures below about 600 °C (Walrafen et al. 1999, Foustoukos and Mysen 2012). Linkage of OH groups to Na+ lowers the OH fundamental frequency compared with the spectrum of pure H2O (Mysen and Virgo 1986). This could appear as an asymmetry toward lower frequency as observed in the spectra of the ZrO2-NaOH-H2O and ZrO2-SiO2-NaOH-H2O fluid (Fig. 2a). The difference in FWHH in the spectra of NaOH+ SiO2-bearing and SiO2-absent but NaOH-bearing compositions likely reflects the formation of Si-OH linkages in systems with SiO2. This is because Si-OH linkage tends to shift the Raman bands assigned to the fundamental OH vibrations to a higher frequency than that of OH in pure H2O.
The Raman spectra of fluids and crystals in the low-frequency regime characteristically comprise one or more intensity envelopes between 300 and 800 cm−1 (Fig. 4). From the similar Raman spectra of fluid in the ZrO2-H2O and ZrO2-SiO2-H2O systems with two bands between 550 and 650 cm−1, it appears that the structure of dissolved Zr-containing complexes in those fluids resembles one another. These two bands also are at nearly the same frequency as the dominant bands of crystalline ZrO2 at the same temperature and pressure. These latter spectral features resemble those of tetragonal ZrO2 in which the oxygen coordination number is 7 (Keramidas and White 1974, Smith and Newkirk 1965). However, Wilke et al. (2012) interpreted their Zr X-ray near-edge spectra (XANES) to be consistent with eightfold coordinated Zr4+ in ZrO2-H2O solutions. The present Raman spectra of ZrO2-H2O and ZrO2-SiO2-H2O fluids cannot be used to distinguish between Zr4+ in seven- or eightfold coordination with oxygen with more precision than to state that the short-range structure of Zr4+ dissolved in H2O resembles that of coexisting crystalline ZrO2. Fluids in the ZrO2-SiO2-H2O system also contain dissolved silicate. This is in the form of Qo, as evidenced by the strong band near 770 cm−1 (Fig. 4; see also Zotov and Keppler 2002, Mibe et al. 2008).

Examples of the Raman spectra of fluids and crystals at conditions as indicated on the individual spectra. Panels a-d) show spectra of fluids from systems and under conditions indicated on the individual spectra. Panels (e) and (f) show examples of crystalline, Zfr-bearing materials, and panel (g) shows an example of the spectrum of fluid from the SiO2-H2O system at temperature and pressure indcated. Note the insert in (d), which shows an expanded version of the 500–700 cm−1 frequency range so as to better show the similarity between this region and the same region in the spectra of ZrO2-H2O fluids in (a)
The spectra of NaOH-bearing fluids characteristically have an additional strong band centered near 350 cm−1 and one or two bands near 500 cm−1. These were not observed in the spectra of fluids without NaOH. It is also noted that the spectra of ZrO2-SiO2-NaOH-H2O fluids differ from those of ZrO2-NaOH-H2O in that the bands in the 400–600 cm−1 region of the ZrO2-SiO2-NaOH-H2O spectra are narrower and better defined than those of the ZrO2-NaOH-H2O fluid and that the 650 cm−1 band in the ZrO2-NaOH-H2O fluid spectra is absent in the spectra of ZrO2-SiO2-NaOH-H2O (Fig. 4). The Raman spectra of ZrO2-SiO2-NaOH-H2O fluids are notable in their absence of Raman signals that could be assigned to vibrations in silicate tetrahedra in the structure (e.g., the absence of the Qo species with its diagnostic 770 cm−1 band seen in the spectra of SiO2-H2O and ZrO2-SiO2-H2O fluids; see Fig. 4). In comparison with the Raman spectra of crystalline Zr-compounds, the spectrum of Na-zirconate Na2ZrO3 characteristically has two strong bands near 500 cm−1 and a single strong band near 350 cm−1. The spectrum resembles that of BaZrO3 with a sixfold coordinated Zr4+ (Dixit et al. 2004). Data, mostly X-ray-based, from Zr-bearing silicate glasses have been interpreted to indicate that Zr4+ is in sixfold coordination with oxygen (Farges et al. 1991, Galoisy et al. 1999; Farges and Rossano 2000). From this limited information, we conclude that Zr4+ in ZrO2-NaOH-H2O fluids coexisting with crystalline ZrO2 likely forms zirconate complexes possibly with local structural similarity to crystalline zirconate. This structural interpretation also is consistent with published XANES and EXAFS (Extended X-ray Absorption Fine Structure) spectra of Zr-bearing Na-silicate aqueous fluids, where structures resembling zirconosilicates were proposed (Wilke et al. 2012, Louvel et al. 2013). Alkali zirconosilicate complexing also has been suggested for hydrous, peralkaline aluminosilicate melts (Watson 1979).
The composition control of how Zr4+ can form complexes with other solutes in an aqueous solution in aqueous fluids shows some similarities with the behavior of Ti4+ in aqueous fluids at similar temperatures and pressures. In the TiO2-H2O system, for example, the solubility is in the ppm range under similar temperature and pressure conditions (e.g., Audetat and Keppler 2005), and the local structural environment resembles that of TiO2 with Ti4+ in sixfold coordination with oxygen (Mysen 2012). In Na-silicate-bearing solutions, on the other hand, the Ti4+ solubility is several orders of magnitude greater (Antignano and Manning 2008), and Ti4+ is dissolved in aqueous fluid as TiO4- or TiO3-type complexes with Ti4+ attaining a coordination number near 4 (Mysen 2012).
Solubility
The zirconium concentration in a fluid in equilibrium with crystalline ZrO2 may be estimated by comparing the intensities of Raman bands from the fluid and from the crystalline ZrO2 recorded at the same temperature and pressure. In such a calculation, it is assumed that the integrated Raman intensity of spectra and fluid and melt are related to one another via the same correction factor because the local structure of the Zr complexes in such fluids resembles that of crystalline ZrO2 at the same temperature and pressure. This approach to solubility determination is similar to that for Ti solubility in aqueous fluid in equilibrium with crystalline TiO2 (Mysen 2012) where Ti solubility determined in this manner compared well with values obtained by other methods (Audetat and Keppler 2005, Antignano and Manning 2008).
The Zr4+ concentration in ZrO2-H2O fluid ranges from ~10 ppm at 600 °C/60 MPa to about 40 ppm Zr at 900 °C and 560 MPa (Fig. 5). Two data points from the ZrO2-SiO2-H2O system at 725 °C/783 MPa and 800 °C/~1050 MPa indicate solubilities near 60 ppm or about 25 ppm higher than that in the absence of SiO2. This slightly higher solubility likely is because of the higher pressures in the experiments in the ZrO2-SiO2-H2O system because Zr4+ solubility in aqueous fluids is positively correlated with pressure (Ayers et al. 2012, Wilke et al. 2012).

Zr concentration in fluids as a function of temperature and pressure for the systems indicated. Average pressure is that calculated from the regression line in Fig. 2. For actual pressure, see Table 1. Only two data points (at 725 and 800 °C) are shown for fluids in the ZrO2-SiO2-H2O system because at a lower temperature (and pressure) Raman signals that could be assigned to vibrations in Zr-bearing complexes in the fluid were not detected above background noise. Horizontal error bars represent calculated pressure uncertainty (±110 MPa; see text)
The Zr4+ concentration in aqueous fluid is sensitive to alkali and alkali + silica content. For example, addition of 1 M NaOH to H2O raises the Zr4+ content of the aqueous fluid by approximately a factor of 10. Addition of SiO2 to NaOH-bearing systems increases the solubility by an additional factor of 5 or more (Fig. 5). Addition of Na-silicate component also results in major zircon (ZrSiO4) solubility increases (Ayers et al. 2012, Wilke et al. 2012). This feature may be analogous to the solution behavior of Zr4+ in hydrous and peralkaline aluminosilicate melt where the solubility is a positively correlated peralkalinity of the melt (Watson 1979). This concept would also be consistent with the XANES data on silicate glasses by Farges and Rossano (2000). We note, however, that addition of SiO2 to ZrO2-H2O without NaOH does not have significant influence on the Zr4+ solubility. The solubility increase with NaOH qualitatively is similar to that reported elsewhere for zircon (ZrSiO4) and for ZrO2 solubility in aqueous fluid (e.g., Wilke et al. 2012). However, in order to quantitatively compare Zr4+ concentration in fluid equilibrated with ZrO2 from the experiments by Wilke et al. (2012), their data were extrapolated linearly to the temperature/pressure conditions of the present experiments. The resulting solubility compares well with the results reported here (Fig. 5).
Discussion
It has been suggested that pH is an important variable governing Zr solubility in aqueous fluid based on the positive correlation of Zr4+ solubility with NaOH concentration in the fluid. The formation of hydroxylated Zr complexes such as Zr(OH)3+, Zr(OH)2 2+, Zr(OH)3 3+, Zr(OH)4, and Zr(OH)5 − could then be an important mechanism by which Zr4+ can dissolve in aqueous fluid (e.g., Ayers et al. 2012). The interpretation of the Raman signals assigned to fundamental OH vibrations in the 3000–4100 cm−1 frequency range from fluids in the ZrO2-H2O and ZrO2-SiO2-H2O systems does not require the existence of Zr-OH complexes in the fluids as there is no clear evidence for Zr-OH bonding in the spectra. However, crystalline Zr(OH)4 exists, so one cannot rule out undetected OH-bearing Zr complexes. That caveat notwithstanding, for these simple ZrO2-H2O and ZrO2-SiO2-H2O systems, a simple solution mechanism is proposed:
with the associated equilibrium constant:
Here, mZrO2(fluid) is the ZrO2 molality of the fluid. The mZrO2(fluid) was calculated from the weight fraction of ZrO2 according to the expression:
where M subscript denotes molecular weight of the subscripted entity, and X wt subscript is the corresponding weight fraction (in g ZrO2/kg H2O) that was derived from the Raman spectra as discussed above.
The temperature-dependent equilibrium constant, K (1), (Fig. 6), yields ∆H = 43 ± 16 kJ/mol (Tables 2 and 3). In these (and succeeding) calculations, it is assumed that the pressure change associated with changing temperature is not a factor. In other words, the ∆V values of the equilibrium reactions are considered negligible.

Temperature-dependence of the equilibrium constant, K (1), for equilibrium (1). Regression coefficients are shown in Tables 2 and 3. Average pressure is that calculated from the regression line in Fig. 2. For actual pressure, see Table 1. Horizontal error bars represent calculated pressure uncertainty (±110 MPa; see text)
The Zr4+ solubility behavior in the ZrO2-SiO2-H2O system appears similar to that in the ZrO2-H2O (Fig. 5). However, only two data points (at 725 and 800 °C) were obtained for the ZrO2-SiO2-H2O system, and these were within 75 °C of each other because a Raman signal above background noise could be detected at lower temperatures. No clear temperature-dependence could be discerned. Given the data uncertainty, ∆H for reaction (1) in the ZrO2-SiO2-H2O system could not be obtained, but with the similar solubility in fluid in the two systems, the Zr4+ solution mechanism and its energetics likely also are similar.
Addition of 1 M NaOH to the aqueous solution results in a Zr4+ solubility increase of approximately an order of magnitude compared with the NaOH-free solution (Fig. 5). This positive correlation between solubility and NaOH concentration of the fluid was why Ayers et al. (2012) suggested that Zr4+ dissolved as hydroxylated Zr complexes. However, enhanced solubility could just as easily be ascribed to Na-zirconate complexing. Na-zirconate type of complexing is consistent with the available XANES data (Louvel et al. 2013). It is also consistent with the Raman spectra of solutions and crystalline Na-zirconate discussed above (Fig. 4).
Detailed stoichiometry of the zirconate complex(es) cannot be inferred from existing data. The simplest possible zirconate group is ZrO4 4− so that a complex resembling Na4ZrO4 could be considered. The solution mechanism then can be described with the expression:
with the equilibrium constant:
In equilibrium with pure ZrO2, the ZrO2 activity equals unity. The molality of NaOH also equals 1 so that Eq. (5) reduces to:
The \( {m}_{{\mathrm{Na}}_4{\mathrm{ZrO}}_4} \) is calculated from the solubility weight fraction of Na4ZrO4 by substituting \( {M}_{{\mathrm{Na}}_4{\mathrm{ZrO}}_4} \) and \( {X}_{{\mathrm{Na}}_4{\mathrm{ZrO}}_4}^{\mathrm{wt}} \) for \( {M}_{{\mathrm{ZrO}}_2} \) and \( {X}_{{\mathrm{ZrO}}_2}^{\mathrm{wt}} \) in Eq. (3).
The regression of ln K (4) vs. 1/T for the ZrO2-NaOH-H2O fluids is divided into high density and low density (Fig. 6) because the three highest temperature data points for this composition were recorded at a lower pressure as the result of extensive deformation of the sample hole of the Ir gasket (see also Table 1 and Fig. 1). The linear slope of these two curves does agree within error with the ∆H value derived from this temperature-dependence between 195 ± 24 kJ/mol and 232 ± 29 kJ/mol (Fig. 7; Tables 2 and 3). There is, however, an offset of the solubility axis of that figure, which likely reflects the positive correlation of solubility with pressure at any temperature. The K (4) values calculated from the experimental data for ZrO2-NaOH-H2O from Wilke et al. (2012) in the same temperature and pressure range fall nearly within the range of the present data (Fig. 7) with a slightly higher ∆H value (282 kJ/mol; see Tables 2 and 3). It is somewhat difficult to establish the error for those latter data because of the way they were extracted from Wilke et al. (2012) by interpolation and extrapolation of data obtained at different pressure than those of the present experiments.

Temperature-dependence of the equilibrium constant, K (4), for equilibrium (4) for the compositions indicated. Regression coefficients are shown in Tables 2 and 3. Average pressure is that calculated from the regression line in Fig. 2. For actual pressure, see Table 1. Horizontal error bars represent calculated pressure uncertainty (±110 MPa; see text)
The differences in the spectral topology of fluids below 800 cm−1 in (Fig. 4) suggest a different structural behavior of Zr4+ in the ZrO2-NaOH-H2O and ZrO2-SiO2-NaOH-H2O fluids. The 3000–4100 cm−1 intensity envelope also indicates structural differences where at any temperature the FWHH is considerably smaller in spectra of fluids in the ZrO2-SiO2-NaOH-H2O system compared with those in the ZrO2-NaOH-H2O system (Fig. 3). Such narrowing of the Raman band would be consistent with fundamental OH vibrations from OH groups forming Si-OH bonds perhaps linked to Qo species (SiO4 groups). However, the lack of a detectable 770 cm−1 band, which is assignable to protonated orthosilicate (SiO4) groups (e.g., Mibe et al., 2008), makes that possibility less likely. An Si-OH linkage could, nevertheless, exist as a part a zirconosilicate complex in the fluid. This latter suggestion would be consistent with the EXAFS- and XANES-based structural interpretation of fluids in alkali silicate zirconate environments (Wilke et al. 2012, Louvel et al. 2013).
From existing X-ray data and the Raman data reported here, a number of reactions involving zirconosilicate or separate silicate and zirconate complexes could be considered. These possibilities include, but are not unique, an expression such as:
Given the interpretation of the high-frequency envelope of the fluids in ZrO2-SiO2-NaOH-H2O suggesting some Si-OH bonding, it is possible that a portion of Na+- in Na2Zr2Si2O9 might be replaced with H+, which leads to a possible modification of Eq. (7);
with the equilibrium constant:
In Eq. (8), the activity of SiO2 and ZrO2 equals 1 as these are pure crystalline phases. The m NaOH also equals 1. Therefore, K (7b) can be simplified to:
From the temperature-dependent K (7b), ∆H is 30 ± 4 kJ/mol (Fig. 8; Tables 2 and 3). In other words, the Zr solubility is less temperature-dependent than fluids in SiO2-free environments.

Temperature-dependence of the equilibrium constant, K (7b), for equilibrium (7b). Regression coefficients are shown in Tables 2 and 3. Average pressure is that calculated from the regression line in Fig. 2. For actual pressure, see Table 1. Horizontal error bars represent calculated pressure uncertainty (±110 MPa; see text)
Conclusions
The experimental Zr4+ solubility data for water-rich fluids as a function of other solutes illustrate how an essentially insoluble high field strength cation such as Zr4+ can be made quite soluble in aqueous fluid under deep crustal and upper mantle temperature and pressure conditions through the stabilization of zirconate complexes that incorporate other solutes. This conclusion implies, for example, that fluid released during high-temperature dehydration of hydrous mineral assemblages such as those of oceanic crust descending into the upper mantle in subduction zones may carry significant (perhaps thousands of ppm) Zr and likely other HFSEs. Such fluids will alter the environment through which they migrate. This is consistent with natural evidence for Zr4+ transport such as that observed in veins in high-grade metamorphic eclogite and granulite (e.g., Philippot and Selverstone 1991, Möller et al. 2002). Moreover, aqueous fluids transported from dehydrating oceanic crust can also carry this signature to the source rock of partial melting. These processes may have been operational through for at least the last 2 billion years of the Earth’s history where subduction resembling that observed today was operational. Fluid-mediated HFSE transport likely also occurred earlier in the Earth’s history, but those transport mechanisms are less certain.
Abbreviations
- ∆H:
-
change of enthalpy
- ∆V:
-
change of volume
- CCD:
-
charge-coupled detector
- FWHH:
-
full width at half height
- HDAC:
-
hydrothermal diamond anvil cell
- HFSE:
-
high field strength element
- NSF:
-
National Science Foundation
- XANES:
-
X-ray near-edge spectroscopy
References
Ampian SG. X-ray and optical crystallographic data for Na2ZrO3. J Am Ceram Soc. 1968;51:607–8.
Antignano A, Manning CE. Rutile solubility in H2O, H2O –SiO2, and H2O-NaAlSi 3O8 fluids at 0.7-2.0 GPa and 700–1000 °C; implications for mobility of nominally insoluble elements. Chem Geol. 2008;255:283–93.
Audetat A, Keppler H. Solubility of rutile in subduction zone fluids, as determined by experiments in the hydrothermal diamond anvil cell. Earth Planet Sci Lett. 2005;232:393–402.
Ayers JC, Watson EB. Rutile solubility and mobility in supercritical aqueous fluids. Contrib Mineral Petrol. 1993;112:321–30.
Ayers JC, Zhang L, Luo Y, Peters TJ. Zircon solubility in alkaline aqueous fluids at upper crustal conditions. Geochim Cosmochim Acta. 2012;96:18–28.
Bassett WA, Wu T-C, Chou I-M, Haselton T, Frantz JD, Mysen BO. The hydrothermal diamond anvil cell (DAC) and its applications. In: Dyar MD, McCammon CA, Schaefer M, editors. Mineral spectroscopy: a tribute to Roger G. Burns. Houston: The Geochemical Society; 1996. p. 261–72.
Bebout GE. Metamorphic chemical dynamics of subduction zones. Earth Planet Sci Lett. 2007;260:905–20.
Bernini D, Audetat A, Dolejs D, Keppler H. Zircon solubility in aqueous fluids at high temperatures and pressures. Geochim Cosmochim Acta. 2013;119:178–87.
Brenan JM, Shaw HF, Phinney DL, Ryerson RJ. Rutile-aqueous fluid partitioning of Nb, Ta, Hf, Zr, U and Th: implications for high field strength element depletions in island-arc basalts. Earth Planet Sci Lett. 1994;128:327–39.
Compston W, Pidgeon RT. Jack Hills, evidence of more very old detrital zircons in Western Australia. Nature. 1986;321:766–9.
Dixit A, Majumder SB, Dobal PS, Katiyar RS, Bhalla AS. Phase transition studies of sol–gel deposited barium zirconate titanate thin films. Thin Solid Films. 2004;447–448:284–8.
Ellison AJ, Hess PC. Solution behavior of +4 cations in high silica melts; petrologic and geochemical implications. Contrib Mineral Petrol. 1986;94:343–51.
Farges F, Rossano S. Water in Zr-bearing synthetic and natural glasses. Eur J Mineral. 2000;12:1093–107.
Farges F, Ponader CW, Brown CW, Jr GE. Structural environments of incompatible elements in silicate glass/ melt systems: I. Zirconium at trace levels. Geochim Cosmochim Acta. 1991;55:1563–74.
Foustoukos DI, Mysen BO. D/H isotopic fractionation in the H2-H2O system at supercritical water conditions: composition and hydrogen bonding effects. Geochim Cosmochim Acta. 2012;86:88–102.
Frantz JD, Dubessy J, Mysen BO. An optical cell for Raman spectroscopic studies of supercritical fluids and its applications to the study of water to 500°C and 2000 bar. Chem Geol. 1993;106:9–26.
Galoisy L, Pelegrin E, Arrio ME, Ildefonse P, Calas G. Evidence for 6-coordinated zirconium in inactive nuclear waste glasses. J Am Ceram Soc. 1999;82:2219–24.
Giere R. Zirconolite, allanite, and hoegbomite in a marble skarn from the Bergell contact aureole: implications for mobility of Ti, Zr, and REE. Contrib Mineral Petrol. 1986;93:459–70.
Gill JB. Orogenic andesites and plate tectonics. Berlin: Springer; 1981. p. 391.
Hamilton W. Earth’s first two billion years - the era of internally mobile crust. Geol Soc Am Mem. 2007;200:233–96. doi:10.1130/2007.1200(13).
Hanfland M, Syassen K, Fahy S, Louie SG, Cohen ML. Pressure-dependence of the first-order Raman mode in diamond. Phys Rev B. 1985;31:6896–9.
Hermann J, Spandler C, Hack A, Korsakov AV. Aqueous fluids and hydrous melts in high-pressure and ultra-high pressure rocks: implications for element transfer in subduction zones. Lithos. 2006;92:399–417.
Horita J. Hydrogen isotope analyses of natural waters using an H2-water equilibration method: a special implication to brines. Chem Geol. 1988;72:89–94.
Keramidas VG, White WB. Raman scattering study of the crystallization and phase transformations of ZrO2. J Am Ceram Soc. 1974;57:22–4.
Kröner A, Layer PW. Crust formation and plate motion in the early archean. Science. 1992;256:1405–11.
Le Losq CR, Moretti R, Neuville DR. Water speciation in silicate melts investigated by Raman spectroscopy; implication for volcanic processes. Mineral Mag. 2011;75:1296.
Linnen RL, Keppler H. Melt composition control on Zr/Hf fractionation in magmatic processes. Geochim Cosmochim Acta. 2002;66:3293–302.
Louvel M, Sanchez-Valle C, Malfait WJ, Testemale D, Hazemann J-L. Zr complexation in high pressure fluids and silicate melts and implications for the mobilization of HFSE in subduction zones. Geochim Cosmochim Acta. 2013;104:281–99.
Manning CE. The solubility of quartz in H2O in the lower crust and upper mantle. Geochim Cosmochim Acta. 1994;58:4831–40.
Mibe K, Chou I-M, Bassett W A. In situ Raman spectroscopic investigation of the structure of subduction-zone fluids. J Geophys Res. 2008; doi:10.1029/2007JB005179.
Mojzsis SJ, Harrison TM, Pidgeon RT. Mass transfer in the Earth’s mantle and crust is documented on rocks dating to the first few hundred million years of the Earth’s history. Nature. 2001;409:178–81.
Möller A, O’Brian PJ, Kennedy A, Kröner A. Polyphse zircon in ultrahigh-temperature granulites (Rogaland, SW. Norway): constraints from Pb diffusion in zircon. J Metamorph Geol. 2002;20:727–40.
Mysen BO. High-pressure/-temperature titanium solution mechanisms in silicate-saturated aqueous fluids and hydrous silicate melts. Am Mineral. 2012;97:1241–51.
Mysen BO. Carbon speciation in silicate-C-O-H melt and fluid as a function of redox conditions: an experimental study, in-situ to 1.7 GPa and 900 °C. Am Mineral. 2015;100:872–82.
Mysen BO, Virgo D. Volatiles in silicate melts at high pressure and temperature. 1. Interaction between OH groups and Si4+, Al3+, Ca2+, Na+ and H+. Chem Geol. 1986;57:303–31.
Mysen BO, Yamashita S. Speciation of reduced C-O-H volatiles in coexisting fluids and silicate melts determined in-situ to ~1.4 GPa and 800 °C. Geochim Cosmochim Acta. 2010;74:4577–88.
Newton RC, Manning CE, Hanchar JM, Colasanti CV. Free energy of formation of zircon based on solubility measurements at high temperature and pressure. Am Mineral. 2010;95:52–8.
Pascal ML, Anderson GM. Speciation of Al, Si, and K in supercritical solutions: experimental study and interpretation. Geochim Cosmochim Acta. 1989;53:1843–56.
Philippot P, Selverstone J. Trace-element-rich brines ion eclogitic veins: implications for fluid composition and transport during subduction. Contrib Mineral Petrol. 1991;106:417–30.
Schiferl D, Nicol M, Zaug JM, Sharma SK, Cooney TF, Wang S-Y, et al. The diamond 13C/12C isotope Raman pressure sensor system for high temperature/pressure diamond-anvil cells with reactive samples. J Appl Phys. 1997;82:3256–65.
Scholze H. Zur frage der unterschiedung zwischen H2O-molekulen und OH-gruppen in glasern und mineralen. Naturwissenschaften. 1960;47:226–7.
Smith DK, Newkirk HW. Crystal structure of baddeleyite (monoclinic ZrO2) and its relation to polymorphism of ZrO2. Acta Crystallogr A. 1965;18:983–91.
Tatsumi Y, Eggins S. Subduction zone magmatism. Cambridge: Blackwell Scientific Publications; 1995.
Valley JW, Lackey JS, Cavoisie AJ, Clenchenko CC, Spicuzza MJ, Basel MAS, et al. 4.4 billion years of crustal maturation: oxygen isotope ratios of magmatic zircon. Contrib Mineral Petrol. 2005;150:561–80.
Walrafen GE, Fisher MR, Hokmabadi MS, Yang W-H. Temperature dependence of the low- and high-frequency Raman scattering from liquid water. J Chem Phys. 1986;85:6970–82.
Walrafen GE, Yang W-H, Chu YC. Raman spectra from saturated water vapor to the supercritical fluid. J Chem Phys B. 1999;103:1332–8.
Watson EB. Two-liquid partition coefficients; experimental data and geochemical implications. Contrib Mineral Petrol. 1976;56:119–34.
Watson EB. Zircon saturation in felsic liquids; experimental results and applications to trace element geochemistry. Contrib Mineral Petrol. 1979;70:407–19.
Wilke M, Schmidt C, Dubrail J, Appel K, Borchert M, Kvashnina K, et al. Zircon solubility and zircon complexation in H2O+Na2O+SiO2±Al2O3 fluids at high pressure and temperature. Earth Planet Sci Lett. 2012;349–350:15–25.
Wyllie PJ, Sekine T. The formation of mantle phlogopite in subduction zone hybridization. Contrib Mineral Petrol. 1982;79:375–80.
Zotov N, Keppler H. Silica speciation in aqueous fluids at high pressures and high temperatures. Chem Geol. 2002;184:71–82.
Acknowledgements
Support from NSF grant EAR-1212754 made this research possible. The W. M. Keck Foundation supported acquisition of analytical instruments used. The carbon-13 diamonds were produced by Mr. J. Lai. His assistance is gratefully acknowledged. All data used in this manuscript are available from the author upon request.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author declares that he/she has no competing interests.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Mysen, B. An in situ experimental study of Zr4+ transport capacity of water-rich fluids in the temperature and pressure range of the deep crust and upper mantle. Prog. in Earth and Planet. Sci. 2, 38 (2015). https://doi.org/10.1186/s40645-015-0070-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40645-015-0070-5