
Abstract
Background
In vitro reconstitution of an artificial metabolic pathway has emerged as an alternative approach to conventional in vivo fermentation-based bioproduction. Particularly, employment of thermophilic and hyperthermophilic enzymes enables us a simple preparation of highly stable and selective biocatalytic modules and the construction of in vitro metabolic pathways with an excellent operational stability. In this study, we designed and constructed an artificial in vitro metabolic pathway consisting of nine (hyper)thermophilic enzymes and applied it to the conversion of glycerol to lactate. We also assessed the compatibility of the in vitro bioconversion system with methanol, which is a major impurity in crude glycerol released from biodiesel production processes.
Results
The in vitro artificial pathway was designed to balance the intrapathway consumption and regeneration of energy and redox cofactors. All enzymes involved in the in vitro pathway exhibited an acceptable level of stability at high temperature (60°C), and their stability was not markedly affected by the co-existing of up to 100 mM methanol. The one-pot conversion of glycerol to lactate through the in vitro pathway could be achieved in an almost stoichiometric manner, and 14.7 mM lactate could be produced in 7 h. Furthermore, the in vitro bioconversion system exerted almost identical performance in the presence of methanol.
Conclusions
Many thermophilic enzymes exhibit higher stability not only at high temperatures but also in the presence of denaturants such as detergents and organic solvents than their mesophilic counterparts. In this study, compatibilities of thermophilic enzymes with methanol were demonstrated, indicating the potential applicability of in vitro bioconversion systems with thermophilic enzymes in the conversion of crude glycerol to value-added chemicals.
Similar content being viewed by others


1Background
Integration of diverse biocatalytic modules to construct an advanced microbial cell factory has emerged as a powerful approach for the production of industrially important metabolites [1]. Bioprospecting efforts for exploring novel biocatalytic molecules with unique properties have inspired the design and construction of a wider variety of artificial metabolic pathways [2]. However, installation of an artificially engineered metabolic pathway in living organisms often leads to a competition with natural metabolic pathways for intermediates and cofactors, resulting in insufficient yield of desired metabolites. A possible solution to this problem is to avoid the use of living microorganisms and to construct an in vitro artificial metabolic pathway in which only a limited number of enzymes are involved. Until now, a variety of in vitro synthetic pathways have been designed and constructed for the production of alcohols [3],[4], organic acids [5],[6], carbohydrates [7], hydrogen [8],[9], bioplastic [10], and even electricity [11]. Particularly, employment of enzymes derived from thermophiles and hyperthermophiles enables the simple preparation of catalytic modules with excellent selectivity and thermal stability [5],[12]. Furthermore, although the detailed mechanisms remain to be clarified, many thermophilic enzymes have also been reported to display higher tolerance towards denaturants such as detergents and organic solvents than their mesophilic counterparts [13],[14], and activities of some thermophilic enzymes are even improved with organic solvents [15]. These excellent stabilities of thermophilic enzymes allow great flexibility in the operational conditions of in vitro bioconversion systems.
Concerns about the global warming and depletion of fossil fuel reserves have led to the rapid increase of biodiesel production. Generally, 10 kg of crude glycerol, which is the primary byproduct of the biodiesel industry, is released for every 100 kg of biodiesel and the growing production of biodiesel has resulted in a worldwide surplus of crude glycerol [16]. Although many studies have been conducted to use crude glycerol as a starting material for the fermentation-based production of industrially valuable chemicals, these attempts often suffer from the inhibitory effects of impurities contained in crude glycerol on the growth and biocatalytic activity of living organisms [17],[18]. Particularly, methanol, which is the most abundant impurity in crude glycerol, accounts for up to 70% (w/w) of a raw glycerol obtained through a biodiesel production process [19]. In this study, we focused on the high operational stabilities of thermophilic enzymes and employed them as modules to construct an in vitro synthetic pathway for the conversion of glycerol to lactate, which is one of the most important and versatile biomass-derived chemical [20], in the presence of methanol.
2Methods
2.1 Materials
Glycerol and methanol were purchased from Wako Pure Chemical Industries Ltd. (Osaka, Japan). Other intermediates of the synthetic pathway, including glycerol-3-phosphate (G3P), dihydroxyacetone phosphate (DHAP), glyceraldehyde-3-phosphate (GAP), 3-phosphoglycerate (3-PG), 2-phosphoglycerate (2-PG), phosphoenolpyruvate (PEP), and pyruvate were obtained from Sigma-Aldrich Japan (Tokyo, Japan). NAD+, NADH, ADP, and ATP were products of Oriental Yeast Co. Ltd. (Osaka, Japan). 2-(4-Iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt (WST-1), and 1-methoxy-5-methylphenazinium methylsulfate (1-methoxy PMS) were purchased from Dojindo Laboratories (Kumamoto, Japan). All other reagents were commercially available and of analytical grade.
2.2 Microorganisms and plasmid
Escherichia coli JM109 was used for general cloning purpose. E. coli Rosetta 2 (DE3) was used for gene expression. Recombinant E. coli was aerobically cultivated at 37°C in Luria-Bertani medium supplemented with 100 μg ml−1 ampicillin and 34 μg ml−1 chloramphenicol. Gene expression was induced by the addition of 0.2 mM isopropyl β-D-1-thiogalactopyranoside at the late log phase. The expression vector encoding the glycerol kinase of Thermococcus kodakarensis (GKTk, gi| 3986088) was donated by Dr. Y. Koga, Osaka University [21]. Sources of expression vectors for triose phosphate isomerase (TIMTt, gi| 3169211), enolase (ENOTt, gi| 55979971), pyruvate kinase (PKTt, gi| 55979972), lactate dehydrogenase (LDHTt, gi| 55981082) of Thermus thermophilus, non-phosphorylating GAP dehydrogenase of T. kodakarensis (GAPNTk, gi|57640640), and cofactor-independent phosphoglycerate mutase of Pyrococcus horikoshii (iPGMPh, gi|14589995) were described previously [5]. The expression vector for G3P dehydrogenase of T. thermophilus (G3PDHTt, gi|55981709) was obtained from the Riken T. thermophilus HB8 expression plasmid set [22]. Gene encoding NADH oxidase of Thermococcus profundus (NOXTp, gi|187453160) was cloned and expressed in E. coli as described elsewhere [12].
2.3 Analytical method
Lactate was quantified by high-performance liquid chromatography (HPLC) equipped with two tandemly connected ion exclusion columns (Shim-pack SPR-H, 250 mm × 7.8 mm, Shimadzu Corp., Kyoto, Japan). The columns were eluted at 50°C using 4 mM p-toluenesulfonic acid as a mobile phase at a flow rate of 0.2 ml min−1. The eluent was mixed with a pH-buffered solution (16 mM Bis-Tris, 4 mM p-toluenesulfonic acid, and 0.1 mM EDTA) supplied at a flow rate of 0.2 ml min−1 and then analyzed for lactate using a conductivity detector (CDD-20A, Shimadzu Corp.). Methanol concentration was quantified by an enzymatic assay using the alcohol dehydrogenase (Sigma-Aldrich Japan) and the horseradish peroxidase (Sigma-Aldrich Japan) according to the protocol provided by the manufacturer.
2.4 Enzyme assay
E. coli cells were collected by centrifugation, resuspended in 50 mM HEPES-NaOH (pH 7), and then disrupted by a UD-201 ultrasonicator (Kubota Corp., Osaka, Japan). After the removal of cell debris by centrifugation, the cell-free extract was incubated at 70°C for 30 min. The heat-precipitated proteins were removed by centrifugation, and the resulting supernatant was used as an enzyme solution. One unit of an enzyme was defined as the amount consuming 1 μmol of the substrate per min under the below-mentioned standard assay conditions. Protein concentration was measured with the Bio-Rad protein assay kit (Bio-Rad Laboratories Inc., Hercules, CA, USA) using bovine serum albumin as the standard.
Enzyme activities were spectrophotometrically determined at 60°C by monitoring consumption or generation of NADH at 340 nm. When necessary, NADH generation was coupled with the reduction of WST-1 and detected at 438 nm. GKTk activity was determined by coupling with G3PDHTt. The standard assay mixture for GKTk was composed of 50 mM HEPES-NaOH (pH 7), 0.2 mM glycerol, 0.2 mM ATP, 1 mM NAD+, 5 mM MgCl2, 0.5 mM MnCl2, 0.15 mM WST-1, 6 μM 1-methoxy PMS, an excess amount of G3PDHTt, and an appropriate amount of GKTk. The mixture without glycerol was pre-incubated at 60°C for 2 min, and the reaction was initiated by the addition of the substrate. Enzyme reaction was monitored through the reduction of WST-1 to the corresponding formazan dye at 438 nm using a UV-2450 spectrophotometer (Shimadzu Corp.). G3PDHTt assay was performed in the same manner except that 0.2 mM G3P was used as a substrate. TIMTt was assayed in a mixture containing 50 mM HEPES-NaOH (pH 7), 0.2 mM DHAP, 1 mM NAD+, 5 mM MgCl2, 0.5 mM MnCl2, 1 mM glucose-1-phosphate (G1P), an excess amount of GAPNTk, and an appropriate amount of the enzyme. After a pre-incubation at 60°C for 2 min, the substrate was added to the mixture and the reduction of NAD+ was monitored at 340 nm. For the determination of GAPNTk activity, 0.2 mM GAP was used instead of DHAP. Similarly, iPGMPh activity was assessed by coupling with ENOTt, PKTt, and LDHTt. The enzyme was assayed in a mixture containing 50 mM HEPES-NaOH (pH 7), 0.2 mM 3-PG, 0.2 mM ADP, 0.2 mM NADH, 5 mM MgCl2, 0.5 mM MnCl2, and excess amounts of ENOTt, PKTt, and LDHTt. The reaction rate was determined by monitoring the concomitant decrease of NADH at 340 nm. Assays for ENO, PK, and LDH were performed in the same mixture using 0.2 mM each of 2-PG, PEP, and pyruvate, respectively. NOXTp activity was determined by monitoring the oxidation of NADH under an air atmosphere. A reaction mixture comprising 50 mM HEPES-NaOH (pH 7.0), 5 mM MgCl2, 0.5 mM MnCl2, 0.02 mM flavin adenine dinucleotide (FAD), and 0.2 mM NADH was preincubated at 60°C for 2 min and then the reaction was initiated by adding an appropriate amount of enzyme.
2.5 Lactate production
The reaction mixture (4 ml) was composed of 50 mM HEPES-NaOH (pH 7), 0.2 mM glycerol, 1 mM NAD+, 0.2 mM NADH, 0.2 mM ATP, 0.2 mM ADP, 0.02 mM FAD, 0.5 mM FBP, 1 mM G1P, 5 mM MgCl2, and 0.5 mM MnCl2. Enzymes were added to the reaction mixture to give the following final concentrations: 0.04 U ml−1 GKTt, 0.18 U ml−1 G3PDHTt, 0.04 U ml−1 TIMTt, 0.1 U ml−1 GAPNTk, 0.09 U ml−1 iPGMPh, 0.07 U ml−1 ENOTt, 0.09 U ml−1 PKTt, 0.08 U ml−1 LDHTt, and 0.04 U ml−1 NOXTp. The mixture was put in a 10-ml cylindrical vessel and kept at 60°C with stirring. Glycerol (160 mM) solution was continuously supplied to the mixture at a flow rate of 1 μl min−1 (0.04 μmol glycerol ml−1 min−1) using a Shimadzu LC-20 AD solvent delivery unit. Alternatively, a model solution of crude glycerol, which consisted of 160 mM glycerol and 770 mM methanol, was used as a substrate and fed to the reaction mixture in the same manner. NAD+ was put in the substrate solution at 4 mM and supplied into the reaction mixture with the substrate to complement the thermal degradation (0.001 μmol NAD+ ml−1 min−1). Aliquots (50 μl) of the reaction mixture were sampled at every 1-h intervals, diluted fourfold with distilled water. The sample was ultrafiltrated using Amicon 3 K (Merk Milipore, Billerica, MA, USA) and then analyzed by HPLC.
3Results and discussion
3.1 Design of the in vitro synthetic pathway
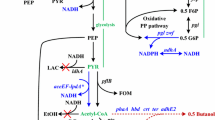
Figure 1 illustrates the newly designed synthetic pathway for the conversion of glycerol to lactate. To construct an in vitro synthetic pathway, it is vital to prevent the depletion of energy and redox cofactors (ATP/ADP, and NAD+/NADH) by balancing their intrapathway consumption and regeneration. In a previous study, we constructed an ATP/ADP-balanced chimeric Embden-Meyerhof (EM) pathway by swapping the enzyme couple of GAP dehydrogenase and phosphoglycerate kinase in the bacterial/eukaryotic EM pathway with the non-phosphorylating GAP dehydrogenase (GAPNTk) involved in the modified EM pathway of a hyperthermophilic archaeon, Thermococcus kodakarensis[5]. Similarly, we employed the GAPN-mediated non-ATP-forming dehydrogenation of GAP to 3-PG for balancing the consumption and regeneration of ATP and ADP through the glycerol converting pathway. On the other hand, the conversion of one molecule of glycerol to lactate through the designed pathway was accompanied by the generation of one molecule of NADH. To re-oxidize the cofactor and to maintain the redox balance of the whole pathway, a hyperthermophilic NADH oxidase was integrated into the pathway. NADH oxidases catalyze the reduction of O2 using NAD(P)H as a reductant and can be divided into two groups: those catalyzing two-electron reduction of O2 to H2O2 and those catalyzing four-electron reduction of O2 to H2O. In this study, we employed the NADH oxidase from T. profundus (NOXTp), which preferably catalyzes four-electron reduction of O2[23], to eliminate the inhibitory effects of H2O2 on enzymes. The chemical equation of the overall reaction through the synthetic pathway can be shown as follows:

Schematic illustration of the in vitro synthetic pathway constructed in this study.
The standard Gibbs energy change (∆G°) of the reaction was calculated to be −256.4 kJ/mol.
3.2 Enzyme stability
Crude extracts of recombinant E. coli cells were heat-treated at 70°C for 30 min to denature indigenous proteins and then used in following studies. SDS-PAGE analysis of the crude extract revealed that most of host-derived proteins were removed by the heat precipitation (Additional file 1: Figure S1). The enzyme stability was assessed by measuring the remaining activity of enzymes after the incubation at 60°C (Figure 2). Most enzymes could retain more than 80% of their initial activity for 8 h, except that PKTt lost 35% of the activity after the incubation for the same time period. We also investigated the effect of methanol, which is the primary impurity contained in crude glycerol, on the enzyme stability. Although residual activities of TIMTt, iPGMPh, ENOTt, and LDHTt were moderately lower than those in the absence of methanol (16% to 32% decrease), the destabilization profile of other enzymes were not significantly affected by at least up to 100 mM of methanol.

Enzyme stability. Enzyme solutions were incubated at 60°C for indicated time periods and residual activities were determined under the standard assay conditions (green circle). Enzyme stabilities were also assessed at 60°C in the presence of 50 (blue circle) and 100 mM methanol (orange circle).
3.3 Optimization of reaction conditions
The lactate production rate through the synthetic pathway was determined at different pH and temperatures by incubating 0.1 U ml−1 each of GKTt, G3PDHTt, TIMTt, GAPNTk, iPGMPh, ENOTt, PKTt, LDHTt, and NOXTp with 10 mM glycerol and appropriate concentrations of cofactors and metal ions (Figure 3). Glucose-1-phosphate (G1P) was put in the reaction mixture as an activator for GAPNTk[24]. Although LDHTt is allosterically inhibited by NAD+[5], lactate dehydrogenases can generally be activated by fructose-1,6-bisphosphate (FBP). In fact, we found that LDHTt activity in the presence of 1 mM NAD+ could be recovered to the similar level to that under the standard assay conditions by the addition of 0.5 mM FBP (Table 1). When the reaction was carried out at 60°C in different buffers, the highest lactate production rate of 0.036 μmol min−1 ml−1 was observed in HEPES-NaOH (pH 7.0) (Figure 3A). The reaction was then performed in this buffer at 50°C, 60°C, and 70°C (Figure 3B). Although no significant difference was observed in production rates at 60°C and 70°C (P >0.1, Student's t-test), the reaction temperature of 60°C was employed for further studies to mitigate the thermal inactivation of the enzymes and the decomposition of thermo-labile intermediates and cofactors.

Effects of pH (A) and temperature (B) on the lactate production through the in vitro synthetic pathway. Enzymes (0.1 U ml−1 each) were incubated in a mixture of 10 mM glycerol, 1 mM NAD+, 0.2 mM NADH, 0.2 mM ATP, 0.2 mM ADP, 0.02 mM FAD, 0.5 mM FBP, 1 mM G1P, 5 mM MgCl2, 0.5 mM MnCl2, and an appropriate buffer. Reaction was performed at indicated pH and temperature for 30 min and then terminated by removing the enzymes with ultrafiltration. Fifty millimolar of MES-NaOH (pH 6) and HEPES-NaOH (pH 7 and 8) were used to adjust pH.
3.4 Lactate production
Unlike highly branched metabolic pathways in living organisms, in vitro synthetic pathways, in which only a limited number of enzyme reactions are sequentially aligned, appear to be less sensitive to the imbalance in enzyme concentrations. Although the existence of a rate-limiting enzyme leads to the accumulation of the specific intermediate, it is eventually converted by downstream enzymes without being routed into the co-existing pathway. However, the accumulation of chemically labile intermediates will result in their spontaneous degradation and decrease in the overall yield of product. We previously demonstrated that the flux through an in vitro metabolic pathway can be spectrophotometrically determined by dividing the whole pathway into some partial pathways, in each of which the NAD(H)-dependent enzymes are assigned to be the last step and by monitoring the concomitant consumption or production of NAD(P)H through the partial pathways [5]. This real-time monitoring technique enables us to identify rate-limiting enzymes in an in vitro pathway by increasing the concentration of each enzyme, one by one. The optimum concentrations of enzymes to achieve a desired flux can be experimentally determined by modulating the concentrations of the rate-limiting enzymes [4],[5]. Accordingly, we divided the glycerol converting pathway in three parts, namely from glycerol to DHAP, from DHAP to 3-PG, and from 3-PG to lactate, and then adjusted the enzyme concentrations in each partial pathway separately. As a result, the optimum enzyme concentrations to achieve a lactate production rate of 0.04 μmol ml−1 min−1 were determined as follows: 0.04 U ml−1 GKTt, 0.18 U ml−1 G3PDHTt, 0.04 U ml−1 TIMTt, 0.1 U ml−1 GAPNTk, 0.09 U ml−1 iPGMPh, 0.07 U ml−1 ENOTt, 0.09 U ml−1 PKTt, and 0.08 U ml−1 LDHTt. Accordingly, NOXTp was put in the reaction mixture to give a NADH re-oxidizing rate of 0.04 μmol ml−1 min−1. A glycerol solution (160 mM) was continuously supplied to the mixture at a rate of 1 μl min−1, which is identical to the experimentally determined lactate production rate through the synthetic pathway, to maintain the pool size of the substrate and a constant flux through the pathway. Although the synthetic pathway was designed to achieve the balanced reduction and oxidation of NAD+ and NADH, the thermal decomposition of the cofactors was not negligible (Additional file 2: Figure S2). Owing to this fact, NAD+ was also continuously supplied to the reaction mixture at a rate identical to that of its thermal decomposition (0.001 μmol min−1 ml−1). The lactate production rate could be remained almost constant at the expected level (0.04 μmol min−1 ml−1) for the initial 5 h (Figure 4). Following this, 11.5 mM lactate could be produced with an overall molar conversion yield of 95.5%. Decrease in the production rate became significant after the initial 5 h, and the conversion yield dropped down to 88% at 7 h. This appeared partly due to the dilution of the reaction mixture (10.5% increase in the total volume at 7 h) caused by the continuous feeding of the substrate solution as well as the loss of enzymes by the sampling (8.8% decrease in the total concentration at 7 h). Decrease in the production rate might also result from the thermal inactivation of PKTt (Figure 2). Substitution of PKTt with another pyruvate kinase derived from hyperthermophiles with higher optimum growth temperature than T. thermophilius may be a possible means for improving the operational stability of the in vitro bioconversion system.

Lactate production through the in vitro synthetic pathway. Production assays were performed using pure glycerol (green circle) and a mixture of glycerol and methanol (orange circle). Total concentration of glycerol fed into the reaction mixture was indicated by a dotted line.
The final lactate concentration after the reaction for 7 h was 14.7 mM. The overall turnover number of ATP/ADP was calculated to be 36.8, while that of NAD+/NADH was 17.5. Enantiomeric purity of the product was determined using a BF-5 biosensor equipped with a D-lactate quantification unit (Oji Scientific Instruments, Amagasaki, Japan). Concentration of D-lactate in the reaction mixture was under the detection limit (approximately 0.05 mM), indicating that glycerol was enantio-specifically converted to L-lactate.
3.5 Compatibility of the in vitro bioconversion system with methanol
The chemical composition of crude glycerol is highly varied with the types of catalysts and feedstocks used for biodiesel production processes [18]. Hansen et al. analyzed the chemical composition of 11 types of crude glycerol obtained from 7 Australian biodiesel manufacturers and revealed that the glycerol content in the crude glycerol varied in the range of 38% to 96% and up to 16.1% of methanol was contained as an impurity [25]. Moreover, Asad-ur-Rehman et al. reported that a raw glycerol obtained during the biodiesel preparation from sunflower oil contained 50% methanol, which is more abundant than the glycerol content (30%) [19]. In order to assess the compatibility of the in vitro system with crude glycerol, a model solution of crude glycerol consisting of 30% (w/v) glycerol and 50% (w/v) methanol was prepared and used as a substrate for lactate production. The model solution was diluted by distilled water to give a final glycerol concentration of 160 mM (thereby the final methanol concentration was 770 mM) and supplied into the reaction mixture in the same manner as the lactate production with pure glycerol. The time profile of the lactate production with the model solution was almost identical to that with pure glycerol (Figure 4). After the reaction for 7 h, methanol concentration in the reaction mixture reached 47.8 mM, which was significantly lower than the calculated concentration of 80.5 mM probably due to volatilization. These results were in reasonable agreement with our observation that stabilities of most enzymes involved in the synthetic pathway were not markedly affected by up to 100 mM of methanol and demonstrated the potential applicability of in vitro bioconversion systems with thermophilic enzymes for the conversion of crude glycerol to value-added chemicals.
4Conclusions
In this study, we constructed an artificial in vitro metabolic pathway for the conversion of glycerol to lactate. The in vitro pathway consisted of nine thermophilic and hyperthermophilic enzymes and designed to balance the intrapathway consumption and regeneration of cofactors. The one-pot conversion of glycerol to lactate through the in vitro pathway could be achieved in an almost stoichiometric manner. Although the final product concentration obtained in this study was modest, the overall yield and the production rate of lactate were comparable to those in conventional fermentation processes [20]. Besides, the in vitro bioconversion system can be operated in a simple buffer solution and thus would markedly simplify downstream processes including product recovery and purification. Furthermore, the in vitro bioconversion system exerted almost identical performance in the presence of methanol, demonstrating the potential of thermophilic-enzyme-based in vitro metabolic engineering approaches in the utilization of crude glycerol as a starting material for the production of value-added chemicals. On the other hand, although their contents are generally less abundant than those of methanol, crude glycerol contains many other impurities, including fatty acids, soap, and salts. Compatibility tests of thermophilic enzymes with these impurities and the implementation of the in vitro bioconversion with a real crude glycerol would be needed in future studies.
Additional files
Abbreviations
- GKTt:
-
glycerol kinase from Thermococcus kodakarensis
- G3PDHTt:
-
glycerol-3-phosphate dehydrogenase from Thermus thermophilus
- TIMTt:
-
triose phosphate isomerase from Thermus thermophilus
- GAPNTk:
-
non-phosphorylating glyceraldehyde-3-phosphate dehydrogenase from Thermococcus kodakarensis
- iPGMPh:
-
cofactor-independent phosphoglycerate mutase from Pyrococcus horikoshii
- ENOTt:
-
enolase from Thermus thermophilus
- PKTt:
-
pyruvate kinase from Thermus thermophilus
- LDHTt:
-
lactate dehydrogenase from Thermus thermophilus
- NOXTp:
-
NADH oxidase from Thermococcus profundus
- G3P:
-
glycerol-3-phosphate
- DHAP:
-
dihydroxyacetone phosphate
- GAP:
-
glyceraldehyde-3-phosphate
- 3-PG:
-
3-phosphoglycerate
- 2-PG:
-
2-phosphoglycerate
- PEP:
-
phosphoenolpyruvate
- G1P:
-
glucose-1-phosphate
- FBP:
-
fructose-1,6-bisphosphate
- FAD:
-
flavin adenine dinucleotide
- WST-1:
-
2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt
- 1-methoxy PMS:
-
1-methoxy-5-methylphenazinium methylsulfate
- HPLC:
-
high-performance liquid chromatography
References
Rabinovitch-Deere CA, Oliver JWK, Rodriguez GM, Atsumi S: Synthetic biology and metabolic engineering approaches to produce biofuels. Chem Rev 2013, 113: 4611–4632. 10.1021/cr300361t
Bond-Watts BB, Bellerose RJ, Chang MCY: Enzyme mechanism as a kinetic control element for designing synthetic biofuel pathways. Nat Chem Biol 2011, 7: 222–227. 10.1038/nchembio.537
Guterl JK, Garbe D, Carsten J, Steffler F, Sommer B, Reiße S, Philipp A, Haack M, Rühmann B, Koltermann A, Kettling U, Thomas Brück T, Sieber V: Cell-free metabolic engineering: production of chemicals by minimized reaction cascades. ChemSusChem 2012, 5: 2165–2172. 10.1002/cssc.201200365
Krutsakorn B, Honda K, Ye X, Imagawa T, Bei X, Okano K, Ohtake H: In vitro production of n -butanol from glucose. Metab Eng 2013, 20: 84–91. 10.1016/j.ymben.2013.09.006
Ye X, Honda K, Sakai T, Okano K, Omasa T, Hirota R, Kuroda A, Ohtake H: Synthetic metabolic engineering - a novel, simple technology for designing a chimeric metabolic pathway. Microb Cell Fact 2012, 11: 120. 10.1186/1475-2859-11-120
Ye X, Honda K, Morimoto Y, Okano K, Ohtake H: Direct conversion of glucose to malate by synthetic metabolic engineering. J Biotechnol 2013, 164: 34–40. 10.1016/j.jbiotec.2012.11.011
You C, Chen H, Myung S, Sathitsuksanoh N, Ma H, Zhang XZ, Li J, Zhang YHP: Enzymatic transformation of nonfood biomass to starch. Proc Natl Acad Sci U S A 2013, 110: 7182–7187. 10.1073/pnas.1302420110
Woodward J, Orr M, Cordaray K, Greenbaum E: Enzymatic production of biohydrogen. Nature 2000, 405: 1014–1015. 10.1038/35016633
Zhang YHP, Evans BR, Mielenz JR, Hopkins RC, Adams MWW: High-yield hydrogen production from starch and water by a synthetic enzymatic pathway. PLoS ONE 2007, 2: e456. 10.1371/journal.pone.0000456
Opgenorth PH, Korman TP, Bowie JU: A synthetic biochemistry molecular purge valve module that maintains redox balance. Nat Commun 2014, 5: 4113. 10.1038/ncomms5113
Zhu Z, Tam TK, Sun F, You C, Zhang YHP: A high-energy-density sugar biobattery based on a synthetic enzymatic pathway. Nat Commun 2014, 5: 3026.
Ninn PH, Honda K, Sakai T, Okano K, Ohtake H (2014) Assembly and multiple gene expression of thermophilic enzymes in Escherichia coli for in vitro metabolic engineering. Biotechnol Bioeng:(in press)
Owusu RK, Cowan DA: Correlation between microbial protein thermostability and resistance to denaturation in aqueous: organic solvent two-phase systems. Enzyme Microb Technol 1989, 11: 568–574. 10.1016/0141-0229(89)90084-7
Atomi H: Recent progress towards the application of hyperthermophiles and their enzymes. Curr Opin Chem Biol 2005, 9: 166–173. 10.1016/j.cbpa.2005.02.013
Pennacchio A, Pucci B, Secundo F, La Cara F, Rossi M, Raia CA: Purification and characterization of a novel recombinant highly enantioselective short-chain NAD(H)-dependent alcohol dehydrogenase from Thermus thermophilus . Appl Environ Microbiol 2008, 74: 3949–3958. 10.1128/AEM.00217-08
Nguyen AQ, Kim YG, Kim SB, Kim CJ: Improved tolerance of recombinant Escherichia coli to the toxicity of crude glycerol by overexpressing trehalose biosynthetic genes ( otsBA ) for the production of β-carotene. Bioresour Technol 2013, 143: 531–537. 10.1016/j.biortech.2013.06.034
Venkataramanan KP, Boatman JJ, Kurniawan Y, Taconi KA, Bothun GD, Scholz C: Impact of impurities in biodiesel-derived crude glycerol on the fermentation by Clostridium pasteurianum ATCC 6013. Appl Microbiol Biotechnol 2012, 93: 1325–1335. 10.1007/s00253-011-3766-5
Yang F, Hanna MA, Sun R: Value-added uses for crude glycerol - a byproduct of biodiesel production. Biotechnol Biofuels 2012, 5: 13. 10.1186/1754-6834-5-13
Asad-ur-Rehman SWRG, Nomura N, Sato S, Matsumura M: Pretreatment and utilization of raw glycerol from sunflower oil biodiesel for growth and 1,3-propanediol production by Clostridium butyricum . J Chem Technol Biotechnol 2008, 83: 1072–1080. 10.1002/jctb.1917
Okano K, Tanaka T, Ogino C, Fukuda H, Kondo A: Biotechnological production of enantiomeric pure lactic acid from renewable resources: recent achievements, perspectives, and limits. Appl Microbiol Biotechnol 2010, 85: 413–423. 10.1007/s00253-009-2280-5
Koga Y, Haruki M, Morikawa M, Kanaya S: Stability of chimeras of hyperthermophilic and mesophilic glycerol kinases constructed by DNA shuffling. J Biosci Bioeng 2001, 91: 551–556. 10.1016/S1389-1723(01)80172-9
Yokoyama S, Matsuo Y, Hirota H, Kigawa T, Shirouzu M, Kuroda Y, Kurumizaka H, Kawaguchi S, Ito Y, Shibata T, Kainosho M, Nishimura Y, Inoue Y, Kuramitsu S: Structural genomics projects in Japan. Nat Struct Biol 2000, 7: 943–945. 10.1038/80712
Jia B, Park S-C, Lee S, Pham BP, Yu R, Le TL, Han SW, Yang J-K, Choi M-S, Baumeister W, Cheong G-W: Hexameric ring structure of a thermophilic archaeon NADH oxidase that produces predominantly H 2 O. FEBS J 2008, 275: 5355–5366. 10.1111/j.1742-4658.2008.06665.x
Matsubara K, Yokooji Y, Atomi H, Imanaka T: Biochemical and genetic characterization of the three metabolic routes in Thermococcus kodakarensis linking glyceraldehyde 3-phosphate and 3-phosphoglycerate. Mol Microbiol 2011, 81: 1300–1312. 10.1111/j.1365-2958.2011.07762.x
Hansen CF, Hernandez A, Mullan BP, Moore K, Trezona-Murray M, King RH, Pluske JR: A chemical analysis of samples of crude glycerol from the production of biodiesel in Australia, and the effects of feeding crude glycerol to growing-finishing pigs on performance, plasma metabolites and meat quality at slaughter. Anim Prod Sci 2009,49(1):54–161.
Acknowledgements
This work was supported in part by the Japan Science and Technology Agency, PRESTO/CREST program. This work was also partly supported by the Japan Society for the Promotion of Science, KAKENHI Grant (26450088). CJ was supported by Japanese Funds-in-Trust, UNESCO Biotechnology School in Asia program and Graduate School Thesis Grant, Chulalongkorn University.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contribution
CJ performed the experiments and wrote the manuscript. SCN supervised the work and wrote the manuscript. MC co-performed the experiments, particularly on the enzyme stability. KO and HO contributed general advice and resource support, as well as edited the manuscript. KH designed all the experiments and wrote the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
40643_2014_18_MOESM1_ESM.pdf
Additional file 1: Figure S1.: SDS-PAGE analysis of the crude extracts of recombinant E. coli cells. Crude extracts were prepared from approximately 1 mg (wet weight) of the cells overproducing indicated thermophilic enzymes and separated on 12% acrylamide gels before (−) and after (+) heat treatment at 70°C for 30 min. Dual Xtra prestained protein standard (Bio-Rad Laboratories Inc.) was used as a protein marker (lanes indicated by M). (PDF 40 KB)
40643_2014_18_MOESM2_ESM.pdf
Additional file 2: Figure S2.: Thermal decomposition of NAD+ and NADH. A mixture of NAD+ (1 mM, indicated by blue circle) and NADH (0.2 mM, green circle) in 50 mM HEPES-NaOH (pH 7.0) was incubated at 60°C. After the incubation for indicated time periods, residual concentrations of the cofactors were determined by HPLC as described elsewhere (Morimoto et al. 2014). Orange circles indicate the total concentration of NAD+ and NADH. Data represent the averages of triplicate assays. (PDF 12 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Jaturapaktrarak, C., Napathorn, S.C., Cheng, M. et al. In vitro conversion of glycerol to lactate with thermophilic enzymes. Bioresour. Bioprocess. 1, 18 (2014). https://doi.org/10.1186/s40643-014-0018-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40643-014-0018-4