
Abstract
Background
Mechanical ventilation (MV) can cause ventilator-induced lung injury (VILI). The innate immune response mediates this iatrogenic inflammatory condition. The receptor for advanced glycation end products (RAGE) is a multiligand receptor that can amplify immune and inflammatory responses. We hypothesized that RAGE signaling contributes to the pro-inflammatory state induced by MV.
Methods
RAGE expression was analyzed in lung brush and lavage cells obtained from ventilated patients and lung tissue of ventilated mice. Healthy wild-type (WT) and RAGE knockout (KO) mice were ventilated with relatively low (approximately 7.5 ml/kg) or high (approximately 15 ml/kg) tidal volume. Positive end-expiratory pressure was set at 2 cm H2O during both MV strategies. Also, WT and RAGE KO mice with lipopolysaccharide (LPS)-induced lung injury were ventilated with the above described ventilation strategies. In separate experiments, the contribution of soluble RAGE, a RAGE isoform that may function as a decoy receptor, in ventilated RAGE KO mice was investigated. Lung wet-to-dry ratio, cell and neutrophil influx, cytokine and chemokine concentrations, total protein levels, soluble RAGE, and high-mobility group box 1 (HMGB1) presence in lung lavage fluid were analyzed.
Results
MV was associated with increased RAGE mRNA levels in both human lung brush samples and lung tissue of healthy mice. In healthy high tidal volume-ventilated mice, RAGE deficiency limited inflammatory cell influx. Other VILI parameters were not affected. In our second set of experiments where we compared RAGE KO and WT mice in a 2-hit model, we observed higher pulmonary cytokine and chemokine levels in RAGE KO mice undergoing LPS/high tidal volume MV as compared to WT mice. Third, in WT mice undergoing the LPS/high tidal volume MV, we observed HMGB1 presence in lung lavage fluid. Moreover, MV increased levels of soluble RAGE in lung lavage fluid, with the highest levels found in LPS/high tidal volume-ventilated mice. Administration of soluble RAGE to LPS/high tidal volume-ventilated RAGE KO mice attenuated the production of inflammatory mediators.
Conclusions
RAGE was not a crucial contributor to the pro-inflammatory state induced by MV. However, the presence of sRAGE limited the production of pro-inflammatory mediators in our 2-hit model of LPS and high tidal volume MV.
Similar content being viewed by others

Background
Mechanical ventilation (MV) is a crucial intervention in the management of the critically ill but can aggravate lung injury, also known as ventilator-associated lung injury (VALI) in patients and termed ventilator-induced lung injury (VILI) in animal models [1]–[5]. The exact molecular mechanisms involved in VILI pathogenesis are incompletely understood, but accumulating evidence indicates that MV triggers an inflammatory response in which innate immunity plays a central role [6]–[11].
Pattern recognition receptors (PRRs) are activated by bacterial products and by damage-associated molecular patterns (DAMPs), which are endogenous molecules released during tissue injury [12]. DAMPs are present in the alveolar compartment during (injurious) MV indicating the potential significance of the DAMP/receptor axis in VILI [7]. The role of some PRRs in VILI has been investigated: VILI is in part mediated by toll-like receptor (TLR) 4 signaling [9],[10]. More recently, the importance of the inflammasome pathway was identified [6],[8]. Less is known about the role of the receptor for advanced glycation end products (RAGE) in VILI.
RAGE is highly expressed in the lungs, primarily on the basolateral membrane of alveolar type I cells [13] and recognizes a variety of molecules including alarmins such as S100 proteins and high-mobility group box 1 (HMGB1) [14],[15]. Soluble RAGE (sRAGE) is a RAGE isoform that lacks the transmembrane and cytosolic part. sRAGE levels can be used as biomarker for alveolar epithelial type I cell injury [16],[17]. sRAGE itself may also influence inflammation as it can compete with cell-surface RAGE for ligand engagement [17]. Studies indicated that RAGE ligands are present in the pulmonary compartment during MV: (1) Long-term MV in patients without acute lung injury increased HMGB1 levels in bronchoalveolar lavage fluid (BALF) [18], (2) 4 h of injurious MV in rabbits induced a fivefold increase of HMGB1 levels in BALF and blocking HMGB1 attenuated VILI [19], and (3) S100A12 and S100A8/A9, members of the S100 family of proteins, are found in BALF of patients with acute respiratory distress syndrome (ARDS) [20],[21]. RAGE-ligand interaction activates intracellular pathways and induces pro-inflammatory cytokines, proteases, and oxidative stress [17].
We hypothesized that RAGE signaling contributes to the pro-inflammatory state induced by MV. For this, we analyzed the expression of RAGE mRNA in lung brush cells and BALF cells obtained from ventilated patients. In addition, we ventilated wild-type (WT) and RAGE knockout (KO) mice, healthy and with lipopolysaccharide (LPS)-induced lung injury, to study the role of RAGE in VILI. Furthermore, the contribution of soluble RAGE was investigated. To establish the presence of VILI, we analyzed alveolar capillary permeability and the pulmonary inflammatory response.
Methods
A more detailed description of the methods is provided in Additional file 1.
Patients
Samples obtained from a previous trial in which patients were randomized to two ventilation strategies during elective surgery were used [22]. The Medical Ethics Committee of the University of Amsterdam approved the study protocol, and informed consent was obtained from all patients [22]. mRNA expression levels of RAGE and hypoxanthine-guanine phosphoribosyl transferase (HPRT) in lung brush samples and BALF cells were determined. Analysis included samples in which RAGE and HPRT were both measurable at both time points allowing paired measurements. The previous trial was not powered to find differences in mRNA levels between the two ventilation groups. We therefore combined the samples from both ventilation strategies.
Mice
The Animal Care and Use Committee of the Academic Medical Center approved all experiments. Eight- to ten-week-old male RAGE KO mice were generated as described previously [23], backcrossed ten times to a C57Bl/6 background, and bred in the animal facility of the Academic Medical Center (Amsterdam, the Netherlands). C57BL/6 age matched WT mice were purchased from Harlan Laboratories B.V. (Horst, the Netherlands).
Design
To obtain a first insight into the role of RAGE in MV-induced inflammation and injury in lungs without pre-existing injury, we randomized healthy WT and RAGE KO mice to 5 h of MV or to a non-ventilated control group. Mice of the MV group were pressure-controlled ventilated with either an inspiratory pressure of 10 cm H2O (VT ~ 7.5 ml/kg) (LVT) or an inspiratory pressure of 18 cm H2O (VT ~ 15 ml/kg) (HVT) (n = 6 to 9/group). Respiratory rate was set at 110 breaths/min in the LVT group and 70 breaths/min in the HVT group, positive end-expiratory pressure was set at 2 cm H2O during both MV strategies. The physiological characteristics of the VILI model used were published in detail previously [24].
Since it has been shown that lungs with pre-existing injury are more susceptible to the effects of MV [25],[26], we randomized in a second set of experiments WT and KO mice with pre-injured lungs (induced by inhalation of 5 μg LPS (Escherichia coli L4130, Sigma Aldrich, St. Louis, MO, USA) 1 h before randomization) to the above described ventilation strategies or spontaneously breathing for 5 h.
In a third set of experiments, we analyzed the presence of sRAGE and HMGB1 in our VILI models. In addition, KO mice with LPS-induced lung injury received 50 μg murine sRAGE or vehicle (saline) intratracheally at the start of HVT MV. Recombinant murine his-tagged sRAGE was a kind gift from P. Nawroth. Sample harvesting and processing were done as described previously [8],[24].
Assays
Total protein was determined using Bradford Protein Assay Kit (OZ Biosciences, Marseille, France). Interleukin (IL)-6, keratinocyte-derived chemokine (KC), macrophage inflammatory protein (MIP)-2, IL-1β, and tumor necrosis factor (TNF)-α levels were measured by enzyme-linked immunosorbent assay (ELISA) (R&D Systems Inc., Minneapolis, MN, USA). sRAGE was measured by Mouse RAGE Duo set ELISA (R&D Systems Inc.) as described before [27]. HMGB1 levels were analyzed by Western blot. Proteins were separated using polyacrylamide gel electrophoresis (Criterion Bis-Tris Precast Gel, Carlsbad, CA, USA); to detect HMGB1, a rabbit polyclonal antibody was used (Abcam Biochemicals, Cambridge, UK). Lung tissue homogenate was used to analyze RAGE and HPRT mRNA levels. Methods were used as described previously [8]. RAGE was stained on paraffin-embedded lung tissue. For this, we used goat anti-mouse RAGE polyclonal antibodies (Neuromics, Edina, MN, USA). Alveolar and bronchial epithelium and vascular endothelium were scored for RAGE presence on a scale 0 (negative) to 3 (very intense); total RAGE staining score represents the sum of all scores.
Statistical analysis
Data represent mean ± SEM. Human samples were analyzed by paired t test or Wilcoxon signed-rank test. One-way analysis of variance with Bonferroni or a Kruskall-Wallis test with Mann-Whitney U as post hoc analysis was used to analyze multiple groups. To compare two groups, a t test or Mann-Whitney U test was used. p < 0.05 was considered statistically significant.
Results
RAGE expression in human lung brush cells is enhanced during MV
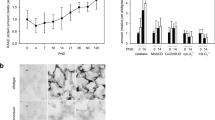
Baseline characteristics, peri-operative parameters and characteristics of the patients included in this study were described in detail previously [22]. BALF cells consisted for more than 99% of macrophages. RAGE mRNA expression levels in these cells (n = 31 pairs) tended to be higher after 5 h of MV, but this did not reach statistical significance (p = 0.12) (Figure 1). In lung brush samples, RAGE mRNA levels were increased after 5 h of MV (n = 15 pairs) (Figure 1). In separate analysis of the two ventilation strategies, no significant differences were found.

RAGE gene expression in ventilated patients. Relative mRNA expression levels of the receptor for advanced glycation end products (RAGE) in human bronchoalveolar lavage fluid cells (A) (n = 31 pairs) and lung brush samples (B) (n = 15 pairs). Samples were obtained from patients at baseline and after 5 h of mechanical ventilation (MV). Gene expression was normalized to the house-keeping gene hypoxanthine-guanine phosphoribosyl transferase (HPRT). Data represent mean (SEM), *p < 0.05.
MV enhanced RAGE expression in healthy murine lungs
To study if MV in healthy mice increases pulmonary RAGE as well, we determined relative RAGE mRNA expression levels in lung tissue homogenates. Both LVT and HVT ventilation strategies significantly increased RAGE mRNA expression in lung tissue when compared to non-ventilated controls (Figure 2). In addition, we analyzed the expression of RAGE on lung immunohistochemical stainings. Whereas RAGE KO lungs did not show any positive staining, healthy non-ventilated control mice abundantly expressed RAGE in their lungs, predominantly in the alveolar epithelium (Figure 2). The total score for RAGE staining was not affected by LVT ventilation but increased in HVT MV. This was mainly due to de novo expression of RAGE on bronchial epithelial cells and endothelium.

RAGE expression in 1-hit VILI. Expression of the receptor of advanced glycation end products (RAGE) in wild-type mice ventilated for 5 h with low tidal volumes (LVT ~ 7.5 ml/kg) or high tidal volumes (HVT ~ 15 ml/kg). Non-ventilated mice (C) served as control. RAGE gene expression was measured in lung homogenates and normalized to the house-keeping gene hypoxanthine-guanine phosphoribosyl transferase (HPRT) (n = 5 for controls, n = 7 ventilated mice/group) (A). Total scores for RAGE expression by immunohistochemical staining of lung tissue (n = 6 for controls, n = 9 ventilated mice/group) (B to E). Representative view of a lung from non-ventilated control mouse (B), absence of RAGE positivity in the lung of a RAGE KO mouse (C), the lung of a LVT-ventilated mouse (D), and increased RAGE expression in the lung of a HVT-ventilated mouse (E). Data represent mean (SEM), *p < 0.05, ***p < 0.001 vs LVT and C, staining magnification ×200.
Neutrophil influx into the alveolar compartment is attenuated in RAGE KO mice in 1-hit VILI
In our 1-hit VILI model, we observed that WT mice subjected to HVT MV demonstrated a marked increase in lung wet-to-dry ratio and total protein level in BALF as compared controls (Figure 3). Further BALF examination demonstrated that both ventilation strategies induced cell influx into the alveolar compartment, with higher levels in HVT-ventilated mice. Differential cell counts revealed an increased number of neutrophils, reaching statistical significance for HVT-ventilated mice. In addition, HVT MV significantly increased the levels of IL-6, KC, and IL-1β. LVT MV elevated the levels of IL-1β and MIP-2 (Table 1). TNF-α was undetectable. When comparing RAGE KO and WT mice, we observed no significant differences regarding alveolar barrier dysfunction. However, RAGE KO mice demonstrated a significantly lower total cell count and neutrophil influx in BALF after 5 h of HVT MV as compared to WT mice (Figure 3). No differences in concentrations of these inflammatory mediators were found between RAGE KO and WT mice (Table 1).

RAGE in 1-hit VILI. Lung wet-to-dry ratios (A) and total protein level (B), cell influx (C), and neutrophil counts (D) in bronchoalveolar lavage fluid of wild-type (WT) and RAGE knockout (KO) mice with healthy lungs ventilated for 5 h with low tidal volumes (LVT ~ 7.5 ml/kg) or high tidal volumes (HVT ~ 15 ml/kg). Non-ventilated mice (C) served as control. Data represent mean (SEM) of n = 6 to 9 mice/group. *p < 0.05, **p < 0.01, and ***p < 0.001.
RAGE KO mice have elevated levels of pro-inflammatory mediators in 2-hit VILI
MV enhanced LPS-induced lung injury in our 2-hit VILI model, most clearly demonstrated in HVT-ventilated mice (Figure 4). When comparing RAGE KO and WT mice, we observed that RAGE deficiency did not affect lung wet-to-dry ratio, total protein level, and cell influx in LPS-exposed and LPS/MV-treated groups (Additional file 2). Also, neutrophil influx was not significantly different between WT and RAGE KO mice (Figure 4). Remarkably, in our 2-hit setting, RAGE KO mice demonstrated elevated cytokine and chemokine levels in BALF. IL-6, TNF-α, and KC were higher when LPS-induced lung injury was combined with MV, reaching significance for the RAGE KO mice of the LPS/HVT MV group (Figure 4). Of note, MIP-2 and IL-1β levels were not different (Additional file 2).

RAGE in 2-hit VILI. Neutrophil counts (A) and levels of interleukin (IL)-6 (B), keratinocyte-derived chemokine (KC) (C) and tumor necrosis factor (TNF)-α (D) in bronchoalveolar lavage fluid of wild-type (WT) and RAGE knockout (KO) mice in a 2-hit lung injury model of lipopolysaccharide (LPS) exposure followed by mechanical ventilation for 5 h with low tidal volumes (LVT ~ 7.5 ml/kg) or high tidal volumes (HVT ~ 15 ml/kg). LPS-exposed non-ventilated mice (C) served as control. Data represent mean (SEM) of n = 6 to 9 mice/group. *p < 0.05, **p < 0.01, and ***p < 0.001.
sRAGE attenuated inflammation in RAGE KO mice
Next, we measured RAGE ligand HMGB1 and sRAGE levels in our VILI models. HVT MV of healthy mice resulted in an increased BALF sRAGE concentration compared to LVT MV and non-ventilated controls (Figure 5). Levels further increased during the LPS/MV double hit in which the highest levels were found in the HVT-ventilated group. HMGB1 was undetectable in 1-hit HVT-ventilated mice. In our 2-hit model, we observed that in lung lavage of animals with LPS-induced lung injury or LPS-induced injury combined with LVT MV, HMGB1 was barely detectable (Figure 6). However, HMGB1 was clearly present in BALF of mice that underwent the LPS/HVT MV double hit (Figure 6). We next verified whether sRAGE might have protective effects during MV by administering sRAGE (or vehicle) to LPS/HVT-ventilated RAGE KO mice. We observed that total protein, cell count, and neutrophil influx were not affected by sRAGE administration (Table 2). However, when analyzing cytokine and chemokine levels in BALF, we found that sRAGE-treated mice displayed significantly lower levels of IL-6, KC, and MIP-2 (Table 2).

sRAGE in lung lavage fluid in 1-hit VILI. Soluble RAGE (sRAGE) levels in bronchoalveolar lavage fluid (BALF) of wild-type (WT) mice ventilated for 5 h with low tidal volumes (LVT ~ 7.5 ml/kg) or high tidal volumes (HVT ~ 15 ml/kg) (VILI) (A). In addition, sRAGE (B) in BALF of WT mice in a 2-hit model of lipopolysaccharide (LPS) exposure followed by mechanical ventilation for 5 h with low tidal volumes (LVT ~ 7.5 ml/kg) or high tidal volumes (HVT ~ 15 ml/kg). Data represent mean (SEM) of n = 6 to 9 mice/group. **p < 0.01, and ***p < 0.001.

HMGB1 in lung lavage fluid in 2-hit VILI. High mobility group box-1 (HMGB1) levels in bronchoalveolar lavage fluid (BALF) of wild-type mice in a 2-hit model of lipopolysaccharide (LPS) exposure followed by spontaneous breathing or mechanical ventilation for 5 h with low tidal volumes (LVT ~ 7.5 ml/kg) or high tidal volumes (HVT ~ 15 ml/kg), n = 8 mice/group.
Discussion
We demonstrate the following: (1) RAGE expression is up-regulated during 5 h of MV in human lung brush cells and in murine lungs. (2) RAGE contributes to inflammatory cell influx in 1-hit VILI, but it does not affect other VILI parameters. (3) In a 2-hit VILI setting, RAGE deficiency is not protective; on the contrary, the lack of RAGE resulted in an enhanced inflammatory response. (4) sRAGE administration to RAGE KO mice undergoing the LPS/HVT MV double hit in part reverses this phenotype.
This is the first study that investigated the role of RAGE in the development of MV-induced inflammation and injury. Others have studied RAGE in other lung injury models such as lung fibrosis, hyperoxyia, LPS, and infection [28]–[31]. In line with previous studies, we observed that in healthy lungs RAGE is already abundantly expressed [16],[28],[29]. Our finding that MV increases RAGE expression in mouse lungs extends a previous investigation studying RAGE in hyperoxia-induced lung inflammation: 4 days of hyperoxia elevated pulmonary RAGE as demonstrated by immunostaining, immunoblotting, and real-time polymerase chain reaction (PCR) [29]. In contrast, other sterile lung inflammatory disorders reported no impact on pulmonary RAGE expression (LPS instillation) [16] or reported reduced RAGE levels (in pulmonary fibrosis models) [28],[30]. This might indicate that the presence of a sustained pro-inflammatory stimulus such as hyperoxia and MV has more impact on RAGE expression. In line, also lung infection models, with the continuous presence of bacteria, reported RAGE up-regulation [31].
In our 1-hit VILI model, RAGE deficiency was associated with a reduced total cell and neutrophil influx into the alveolar compartment. RAGE activation may enhance neutrophil migration by increasing the release of inflammatory mediators. However, we observed no differences in BALF cytokine and chemokine levels at the time point investigated. RAGE itself has also been implicated in the regulation of cell migration: it can function as a counter receptor for leukocyte integrins [32]. In vitro studies showed that RAGE can bind the β2 integrin MAC-1 and p150,95 [32]. From these findings, it can be speculated that RAGE-dependent neutrophil adhesion also contributed in our 1-hit VILI model.
Our current analysis revealed no differences in inflammation in pulmonary LPS-exposed RAGE KO and WT mice. These results are in line with a previous report also showing a similar degree of inflammation between RAGE-deficient and WT mice, here 24 h after intratracheal LPS challenge [33]. However, a recent in vitro study reported evidence for RAGE-LPS interaction [34]. This group also demonstrated that RAGE contributes to LPS-induced nuclear factor-κB activation in isolated peritoneal macrophages, while in vivo RAGE KO mice displayed reduced mortality and inflammation after intraperitoneal LPS-induced shock [34]. As such, the importance of the RAGE-LPS interaction seems to vary in different organ tissues; in the lungs, RAGE-LPS signaling appears less important.
RAGE KO mice undergoing the LPS/HVT MV double hit were not protected from VILI. Instead, they displayed more lung inflammation. This is a remarkable finding since previous findings demonstrated that RAGE deficiency led to attenuated inflammation in sterile lung injury models such as bleomycin-induced and hyperoxia-induced lung injury [29],[35]. However, inflammation was evaluated at a much later time point in these studies: several days after the start of the experiment. Possibly, deficiency of RAGE in our MV model might have influenced respiratory mechanics: pulmonary RAGE is also important for adherence of epithelial cells towards the basal membrane [36]. However, a previous study reported no differences in airway and tissue resistance, compliance, and elastance between healthy RAGE KO and WT mice [37]. We repeated our LPS/HVT MV double hit group in a MV model with volume-controlled ventilation, to exclude possible interference of lung mechanics. Again, an increased inflammatory response in RAGE KO mice was observed (data not shown).
Another explanation for our finding is that the lack of sRAGE was unfavorable for the inflammatory response since sRAGE can scavenge ligands that also have the potential to activate other PRRs. HMGB1 for example can activate not only RAGE but also TLR4 [38]. Moreover, TLR4 signaling clearly contributes to VILI development [9],[10],[39]. We observed elevated HMGB1 levels in mice undergoing the LPS/HVT MV double hit, extending a previous study reporting increased HMGB1 levels after 4 h of extremely high tidal volume (30 ml/kg) MV [19]. Blocking HMGB1 in that study improved alveolar barrier dysfunction and limited neutrophil influx and TNF-α release. In addition, S100A8/A8 proteins, which are as well present in our murine VILI model, also have the potential to trigger both RAGE and TLR4 [15],[40]. The observation that sRAGE administration to RAGE KO mice in part reversed the increased inflammation present in RAGE KO mice indeed suggests that certain ligands were sequestered by sRAGE that would have interacted with other PRRs in the absence of sRAGE. If sRAGE administration could also have therapeutic potential in WT mice in our VILI model is an interesting question for future research. However, a recent study demonstrated that neither intratracheal nor intraperitoneal sRAGE treatment affected LPS-induced or E. coli-induced acute pulmonary inflammation, even though HMGB1 was also increased in these models [33]. In contrast, intraperitoneal sRAGE attenuated lung injury in systemically LPS-challenged mice [41].
We chose to ventilate our mice with low or high tidal volume MV as both strategies may reveal relevant information for clinical practice. Low tidal volume MV is widely practiced since the ARDS network group convincingly demonstrated that this reduces morbidity and mortality in ARDS [2]. However, ARDS is a very heterogeneous disease: some lung regions are poorly aerated placing other healthier lung regions at risk for overinflation [42]. It has been shown that even with the use of lung protective ventilator settings one third of the ARDS patients still experience regional tidal hyperinflation [42]. We therefore believe it is still an important translational effort to ventilate animals with higher tidal volumes. In addition, a pulmonary pro-inflammatory state makes the lung more vulnerable to a second hit such as MV [25],[26]. Therefore, to mimic ventilation in the presence of pulmonary co-morbidities, we added an additional injurious stimulus to our MV model. Nonetheless, our 1-hit VILI model is still important to give insights into RAGE signaling in ventilated healthy lungs.
Our study has several limitations. First, we used patient samples from a previous study in which the effect of two ventilation strategies on inflammation and coagulation was analyzed. This study was, however, not powered to find differences in mRNA levels between the two groups. We therefore combined the samples from both ventilation strategies. Second, we used a short-term VILI model to analyze the role of RAGE. Although short-term MV models are commonly used [8]–[11],[24]–[26],[39], as it is technically very difficult to ventilate rodents for days, a long-term MV model would make the translation of results to clinical practice easier. Third, the tracheotomy and the use of anesthesia might also have influenced inflammation in all ventilated animals. It would be ideal to have a control group of anesthetized, tracheotomized spontaneously breathing mice. Unfortunately, this results in hypoventilation, respiratory acidosis, and death.
Conclusions
In conclusion, our data indicate that RAGE is not a crucial pro-inflammatory receptor in the development of MV-induced inflammation. However, the presence of sRAGE limited the production of pro-inflammatory mediators in ventilated diseased RAGE KO lungs. Further research is needed to study possible therapeutic potential of sRAGE in VILI.
Additional files
Abbreviations
- ARDS:
-
acute respiratory distress syndrome
- BALF:
-
bronchoalveolar lavage fluid
- DAMPs:
-
damage-associated molecular patterns
- ELISA:
-
enzyme-linked immunosorbent assay
- HMGB1:
-
high-mobility group box 1
- HPRT:
-
hypoxanthine-guanine phosphoribosyl transferase
- IL:
-
interleukin
- KC:
-
keratinocyte-derived chemokine
- KO:
-
knockout
- LPS:
-
lipopolysaccharide
- MIP:
-
macrophage inflammatory protein
- MV:
-
mechanical ventilation
- PEEP:
-
positive end-expiratory pressure
- PRRs:
-
pattern recognition receptors
- RAGE:
-
receptor for advanced glycation end products
- sRAGE:
-
soluble RAGE
- TLR:
-
toll-like receptor
- TNF:
-
tumor necrosis factor
- VILI:
-
ventilator-induced lung injury
- WT:
-
wild type
References
Matthay MA, Ware LB, Zimmerman GA: The acute respiratory distress syndrome. J Clin Invest 2012, 122: 2731–2740. 10.1172/JCI60331
Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome N Engl J Med 2000, 342: 1301–1308. 10.1056/NEJM200005043421801
Determann RM, Royakkers A, Wolthuis EK, Vlaar AP, Choi G, Paulus F, Hofstra JJ, de Graaff MJ, Korevaar JC, Schultz MJ: Ventilation with lower tidal volumes as compared with conventional tidal volumes for patients without acute lung injury: a preventive randomized controlled trial. Crit Care 2010, 14: R1. 10.1186/cc8230
dos Santos CC, Slutsky AS: The contribution of biophysical lung injury to the development of biotrauma. Annu Rev Physiol 2006, 68: 585–618. 10.1146/annurev.physiol.68.072304.113443
Lymp JF, Afessa B, Hubmayr RD: Ventilator-associated lung injury in patients without acute lung injury at the onset of mechanical ventilation. Crit Care Med 2004, 32: 1817–1824. 10.1097/01.CCM.0000133019.52531.30
Dolinay T, Kim YS, Howrylak J, Hunninghake GM, An CH, Fredenburgh L, Massaro AF, Rogers A, Gazourian L, Nakahira K, Haspel JA, Landazury R, Eppanapally S, Christie JD, Meyer NJ, Ware LB, Christiani DC, Ryter SW, Baron RM, Choi AM: Inflammasome-regulated cytokines are critical mediators of acute lung injury. Am J Respir Crit Care Med 2012, 185: 1225–1234. 10.1164/rccm.201201-0003OC
Kuipers MT, van der Poll T, Schultz MJ, Wieland CW: Bench-to-bedside review: damage-associated molecular patterns in the onset of ventilator-induced lung injury. Crit Care 2011, 15: 235. 10.1186/cc10437
Kuipers MT, Aslami H, Janczy JR, van der Sluijs KF, Vlaar AP, Wolthuis EK, Choi G, Roelofs JJ, Flavell RA, Sutterwala FS, Bresser P, Leemans JC, van der Poll T, Schultz MJ, Wieland CW: Ventilator-induced lung injury is mediated by the NLRP3 inflammasome. Anesthesiology 2012, 116: 1104–1115. 10.1097/ALN.0b013e3182518bc0
Li H, Su X, Yan X, Wasserloos K, Chao W, Kaynar AM, Liu ZQ, Leikauf GD, Pitt BR, Zhang LM: Toll-like receptor 4-myeloid differentiation factor 88 signaling contributes to ventilator-induced lung injury in mice. Anesthesiology 2010, 113: 619–629. 10.1097/ALN.0b013e3181eaaef9
Vaneker M, Joosten LA, Heunks LM, Snijdelaar DG, Halbertsma FJ, van EJ, Netea MG, van der Hoeven JG, Scheffer GJ: Low-tidal-volume mechanical ventilation induces a toll-like receptor 4-dependent inflammatory response in healthy mice. Anesthesiology 2008, 109: 465–472. 10.1097/ALN.0b013e318182aef1
Villar J, Cabrera NE, Casula M, Flores C, Valladares F, Diaz-Flores L, Muros M, Slutsky AS, Kacmarek RM: Mechanical ventilation modulates TLR4 and IRAK-3 in a non-infectious, ventilator-induced lung injury model. Respir Res 2010, 11: 27. 10.1186/1465-9921-11-27
Bianchi ME: DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol 2007, 81: 1–5. 10.1189/jlb.0306164
Shirasawa M, Fujiwara N, Hirabayashi S, Ohno H, Iida J, Makita K, Hata Y: Receptor for advanced glycation end-products is a marker of type I lung alveolar cells. Genes Cells 2004, 9: 165–174. 10.1111/j.1356-9597.2004.00712.x
Liu R, Mori S, Wake H, Zhang J, Liu K, Izushi Y, Takahashi HK, Peng B, Nishibori M: Establishment of in vitro binding assay of high mobility group box-1 and S100A12 to receptor for advanced glycation endproducts: heparin's effect on binding. Acta Med Okayama 2009, 63: 203–221.
Volz HC, Laohachewin D, Seidel C, Lasitschka F, Keilbach K, Wienbrandt AR, Andrassy J, Bierhaus A, Kaya Z, Katus HA, Andrassy M: S100A8/A9 aggravates post-ischemic heart failure through activation of RAGE-dependent NF-kappaB signaling. Basic Res Cardiol 2012, 107: 1–16. 10.1007/s00395-012-0250-z
Uchida T, Shirasawa M, Ware LB, Kojima K, Hata Y, Makita K, Mednick G, Matthay ZA, Matthay MA: Receptor for advanced glycation end-products is a marker of type I cell injury in acute lung injury. Am J Respir Crit Care Med 2006, 173: 1008–1115. 10.1164/rccm.200509-1477OC
Guo WA, Knight PR, Raghavendran K: The receptor for advanced glycation end products and acute lung injury/acute respiratory distress syndrome. Intensive Care Med 2012, 38: 1588–1598. 10.1007/s00134-012-2624-y
van Zoelen MA, Ishizaka A, Wolthuls EK, Choi G, van der PT, Schultz MJ: Pulmonary levels of high-mobility group box 1 during mechanical ventilation and ventilator-associated pneumonia. Shock 2008, 29: 441–445.
Ogawa EN, Ishizaka A, Tasaka S, Koh H, Ueno H, Amaya F, Ebina M, Yamada S, Funakoshi Y, Soejima J, Moriyama K, Kotani T, Hashimoto S, Morisaki H, Abraham E, Takeda J: Contribution of high-mobility group box-1 to the development of ventilator-induced lung injury. Am J Respir Crit Care Med 2006, 174: 400–407. 10.1164/rccm.200605-699OC
Wittkowski H, Sturrock A, van Zoelen MA, Viemann D, van der Poll T, Hoidal JR, Roth J, Foell D: Neutrophil-derived S100A12 in acute lung injury and respiratory distress syndrome. Crit Care Med 2007, 35: 1369–1375. 10.1097/01.CCM.0000262386.32287.29
Chang DW, Hayashi S, Gharib SA, Vaisar T, King ST, Tsuchiya M, Ruzinski JT, Park DR, Matute-Bello G, Wurfel MM, Bumgarner R, Heinecke JW, Martin TR: Proteomic and computational analysis of bronchoalveolar proteins during the course of the acute respiratory distress syndrome. Am J Respir Crit Care Med 2008, 178: 701–709. 10.1164/rccm.200712-1895OC
Choi G, Wolthuis EK, Bresser P, Levi M, van der Poll T, Dzoljic M, Vroom MB, Schultz MJ: Mechanical ventilation with lower tidal volumes and positive end-expiratory pressure prevents alveolar coagulation in patients without lung injury. Anesthesiology 2006, 105: 689–695. 10.1097/00000542-200610000-00013
Liliensiek B, Weigand MA, Bierhaus A, Nicklas W, Kasper M, Hofer S, Plachky J, Grone HJ, Kurschus FC, Schmidt AM, Yan SD, Martin E, Schleicher E, Stern DM, Hammerling GG, Nawroth PP, Arnold B: Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J Clin Invest 2004, 113: 1641–1650. 10.1172/JCI200418704
Wolthuis EK, Vlaar AP, Choi G, Roelofs JJ, Juffermans NP, Schultz MJ: Mechanical ventilation using non-injurious ventilation settings causes lung injury in the absence of pre-existing lung injury in healthy mice. Crit Care 2009, 13(1):R1. 10.1186/cc7688
Altemeier WA, Matute-Bello G, Gharib SA, Glenny RW, Martin TR, Liles WC: Modulation of lipopolysaccharide-induced gene transcription and promotion of lung injury by mechanical ventilation. J Immunol 2005, 175: 3369–3376. 10.4049/jimmunol.175.5.3369
Dhanireddy S, Altemeier WA, Matute-Bello G, O'Mahony DS, Glenny RW, Martin TR, Liles WC: Mechanical ventilation induces inflammation, lung injury, and extra-pulmonary organ dysfunction in experimental pneumonia. Lab Invest 2006, 86: 790–799. 10.1038/labinvest.3700440
Kalea AZ, Reiniger N, Yang H, Arriero M, Schmidt AM, Hudson BI: Alternative splicing of the murine receptor for advanced glycation end-products (RAGE) gene. FASEB J 2009, 23: 1766–1774. 10.1096/fj.08-117739
Queisser MA, Kouri FM, Konigshoff M, Wygrecka M, Schubert U, Eickelberg O, Preissner KT: Loss of RAGE in pulmonary fibrosis: molecular relations to functional changes in pulmonary cell types. Am J Respir Cell Mol Biol 2008, 39: 337–345. 10.1165/rcmb.2007-0244OC
Reynolds PR, Schmitt RE, Kasteler SD, Sturrock A, Sanders K, Bierhaus A, Nawroth PP, Paine R III, Hoidal JR: Receptors for advanced glycation end-products targeting protect against hyperoxia-induced lung injury in mice. Am J Respir Cell Mol Biol 2010, 42: 545–551. 10.1165/rcmb.2008-0265OC
Englert JM, Hanford LE, Kaminski N, Tobolewski JM, Tan RJ, Fattman CL, Ramsgaard L, Richards TJ, Loutaev I, Nawroth PP, Kasper M, Bierhaus A, Oury TD: A role for the receptor for advanced glycation end products in idiopathic pulmonary fibrosis. Am J Pathol 2008, 172: 583–591. 10.2353/ajpath.2008.070569
van Zoelen MA, Achouiti A, van der Poll T: The role of receptor for advanced glycation endproducts (RAGE) in infection. Crit Care 2011, 15: 208. 10.1186/cc9990
Chavakis T, Bierhaus A, Al-Fakhri N, Schneider D, Witte S, Linn T, Nagashima M, Morser J, Arnold B, Preissner KT, Nawroth PP: The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: a novel pathway for inflammatory cell recruitment. J Exp Med 2003, 198: 1507–1515. 10.1084/jem.20030800
Ramsgaard L, Englert JM, Manni ML, Milutinovic PS, Gefter J, Tobolewski J, Crum L, Coudriet GM, Piganelli J, Zamora R, Vodovotz Y, Enghild JJ, Oury TD: Lack of the receptor for advanced glycation end-products attenuates E. coli pneumonia in mice. PLoS One 2011, 6: e20132. 10.1371/journal.pone.0020132
Yamamoto Y, Harashima A, Saito H, Tsuneyama K, Munesue S, Motoyoshi S, Han D, Watanabe T, Asano M, Takasawa S, Okamoto H, Shimura S, Karasawa T, Yonekura H, Yamamoto H: Septic shock is associated with receptor for advanced glycation end products ligation of LPS. J Immunol 2011, 186: 3248–3257. 10.4049/jimmunol.1002253
He M, Kubo H, Ishizawa K, Hegab AE, Yamamoto Y, Yamamoto H, Yamaya M: The role of the receptor for advanced glycation end-products in lung fibrosis. Am J Physiol Lung Cell Mol Physiol 2007, 293: L1427-L1436. 10.1152/ajplung.00075.2007
Demling N, Ehrhardt C, Kasper M, Laue M, Knels L, Rieber EP: Promotion of cell adherence and spreading: a novel function of RAGE, the highly selective differentiation marker of human alveolar epithelial type I cells. Cell Tissue Res 2006, 323: 475–488. 10.1007/s00441-005-0069-0
Ramsgaard L, Englert JM, Tobolewski J, Tomai L, Fattman CL, Leme AS, Kaynar AM, Shapiro SD, Enghild JJ, Oury TD: The role of the receptor for advanced glycation end-products in a murine model of silicosis. Plos One 2010, 5: e9604. 10.1371/journal.pone.0009604
Andersson U, Tracey KJ: HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol 2011, 29: 139–162. 10.1146/annurev-immunol-030409-101323
Hu G, Malik AB, Minshall RD: Toll-like receptor 4 mediates neutrophil sequestration and lung injury induced by endotoxin and hyperinflation. Crit Care Med 2010, 38: 194–201. 10.1097/CCM.0b013e3181bc7c17
Kuipers MT, Vogl T, Aslami H, Jongsma G, van den Berg E, Vlaar APJ, Roelofs JJTH, Juffermans NP, Schultz MJ, van der Poll T, Roth J, Wieland CW: High levels of S100A8/A9 proteins aggravate ventilator-induced lung injury via TLR4 signaling. Plos One 2013, 8: e68694. 10.1371/journal.pone.0068694
Zhang H, Tasaka S, Shiraishi Y, Fukunaga K, Yamada W, Seki H, Ogawa Y, Miyamoto K, Nakano Y, Hasegawa N, Miyasho T, Maruyama I, Ishizaka A: Role of soluble receptor for advanced glycation end products on endotoxin-induced lung injury. Am J Respir Crit Care Med 2008, 178: 356–362. 10.1164/rccm.200707-1069OC
Terragni PP, Rosboch G, Tealdi A, Corno E, Menaldo E, Davini O, Gandini G, Herrmann P, Mascia L, Quintel M, Slutsky AS, Gattinoni L, Ranieri VM: Tidal hyperinflation during low tidal volume ventilation in acute respiratory distress syndrome. Am J Respir Crit Care Med 2007, 175: 160–166. 10.1164/rccm.200607-915OC
Acknowledgements
We sincerely thank Varun Kumar, Thomas Fleming, and Peter Nawroth from the Department of Internal Medicine I, University Heidelberg, Germany for their generous gift of murine sRAGE. This work was supported by a grant to CW Wieland from The Netherlands Organization for Scientific Research, The Hague, The Netherlands, grant number 91676096.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
MK participated in the study design, performed the experiments, interpreted the work, and drafted the manuscript. HA and PT performed the experimental work, helped interpret the work, and were responsible for the critical review of the manuscript. AT and GJ performed part of the experimental work and interpreted these data (stainings and PCR). KS helped in the analysis of the human samples and interpretation of these data. EW, GC, and PB participated in the study design (human part) and collected the human samples. JR helped with the setup and interpretation of the histological stainings. TP, MS, and CW participated in the study design and interpretation of the data, and drafted the manuscript. All authors critically read and approved the final manuscript.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Kuipers, M.T., Aslami, H., Tuinman, P.R. et al. The receptor for advanced glycation end products in ventilator-induced lung injury. ICMx 2, 22 (2014). https://doi.org/10.1186/s40635-014-0022-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40635-014-0022-1