
Abstract
The canonical Wnt signaling pathway is involved in a variety of biological processes like cell proliferation, cell polarity, and cell fate determination. This pathway has been extensively investigated as its deregulation is linked to different diseases, including various types of cancer, skeletal defects, birth defect disorders (including neural tube defects), metabolic diseases, neurodegenerative disorders and several fibrotic diseases like desmoid tumors. In the "on state", beta-catenin, the key effector of Wnt signaling, enters the nucleus where it binds to the members of the TCF-LEF family of transcription factors and exerts its effect on gene transcription. Disease development can be caused by direct or indirect alterations of the Wnt/β-catenin signaling.
In the first case germline or somatic mutations of the Wnt components are associated to several diseases such as the familial adenomatous polyposis (FAP) - caused by germline mutations of the tumor suppressor adenomatous polyposis coli gene (APC) - and the desmoid-like fibromatosis, a sporadic tumor associated with somatic mutations of the β-catenin gene (CTNNB1).
In the second case, epigenetic modifications and microenvironmental factors have been demonstrated to play a key role in Wnt pathway activation. The natural autocrine Wnt signaling acts through agonists and antagonists competing for the Wnt receptors. Anomalies in this regulation, whichever is their etiology, are an important part in the pathogenesis of Wnt pathway linked diseases. An example is promoter hypermethylation of Wnt antagonists, such as SFRPs, that causes gene silencing preventing their function and consequently leading to the activation of the Wnt pathway. Microenvironmental factors, such as the extracellular matrix, growth factors and inflammatory mediators, represent another type of indirect mechanism that influence Wnt pathway activation. A favorable microenvironment can lead to aberrant fibroblasts activation and accumulation of ECM proteins with subsequent tissue fibrosis that can evolve in fibrotic disease or tumor.
Since the development and progression of several diseases is the outcome of the Wnt pathway cross-talk with other signaling pathways and inflammatory factors, it is important to consider not only direct inhibitors of the Wnt signaling pathway but also inhibitors of microenvironmental factors as promising therapeutic approaches for several tumors of fibrotic origin.
Similar content being viewed by others

Introduction
The Wnt signaling pathway is involved in several essential biological processes in both embryonic development and in adult cell maintenance and regeneration.
The canonical or Wnt/β-catenin dependent pathway controls key developmental gene expression programs by modulating the amount of β-catenin through regulating its degradation or accumulation and its translocation from the adherens junction and cytoplasm to the nucleus. In the absence of a Wnt signal, the cytoplasmic β-catenin is tightly maintained at a low level by a multiprotein destruction complex consisting of Axin, the adenomatous polyposis coli protein (APC), casein kinase 1α (CK1α), and Glycogen Synthase Kinase 3β (GSK3β). The complex phosphorylates cytoplasmic β-catenin leading to its degradation by the ubiquitin-proteasomal system. The continuous elimination of β-catenin prevents its accumulation in the cytoplasm and the consequent translocation into the nucleus. In the presence of a Wnt signal, the destruction complex is disassembled leading to an increment of β-catenin levels and allowing its translocation into the nucleus where it activates Wnt target gene expression. The aberrant regulation of the Wnt/β-catenin pathway plays a role in the pathogenesis of several diseases including cancer, birth defect disorder, skeletal diseases, and fibrotic diseases. For this reason Wnt/β-catenin signaling is tightly regulated and kept under strict control at different levels of the Wnt cascade. Wnt activation is temporally and spatially tuned by autocrine Wnt signaling that is associated with extracellular Wnt agonists and antagonists. The agonists activate the Wnt cascade while the antagonists inhibit Wnt signaling at the level of ligand/receptor [1,2]. However, Wnt/β-catenin signaling deregulation can occur via several mechanisms. In particular, germline mutations of the tumor suppressor gene APC are associated with familial adenomatous polyposis (FAP), and somatic mutations of the β-catenin gene (CTNNB1) are associated with sporadic desmoid tumors. In the first case the disease is caused by a transmissible genetic defect, in the second case the pathology is linked to a somatic mutation that makes β-catenin unable to be completely phosphorylated and degraded.
Wnt/β-catenin signaling can be also indirectly altered by epigenetic modifications that cause silencing of Wnt endogenous brakes, and by the effect of microenvironmental factors, such as the extracellular matrix, hormones and growth factors. Of particular interest is the involvement of inflammatory factors in the modulation of the Wnt/β-catenin pathway and its association with fibrotic disease as well as tumor development.
Either direct or indirect Wnt pathway alterations can cause an increase of β-catenin levels and its accumulation into the nucleus, activating the signaling cascade. The cross-talk between these extracellular stimuli and intracellular signals highlights the complex interaction of the numerous factors involved in the development of the Wnt pathway linked pathologies and are well represented in fibrotic disease and in particular in the sporadic desmoid tumors.
Many studies describe the use of small synthetic molecules for inhibiting the β-catenin as therapeutic approach. Among these, there are molecules that target the interaction of β-catenin with co-activators disabling the formation of an active transcriptional complex. Recently GSK3β inhibitors have been described as promising drugs for several pathologies such as diabetes, stroke, mood disorders, inflammation, and Alzheimer’s disease. The use of specific inhibitors of the Wnt signaling molecules or/and inhibitors of other signaling pathways associated to β-catenin pathway may help to find the key steps of the different pathologies linked to the Wnt pathway.
Review
Wnt pathway
The Wnt pathway is one of the evolutionarily-conserved cell signaling pathways used both during embryogenesis and in developed organism’s homeostasis to regulate cell proliferation, cell polarity, and cell fate determination [3-6]. The extracellular Wnt signal stimulates several intracellular signal transduction cascades, including the non-canonical or β-catenin-independent pathways and the canonical or β-catenin dependent pathway [7].
Non-canonical pathway
The non-canonical Wnt pathways, defined as Wnt- or Frizzled-mediated (Fzd) signaling independent of β-catenin transcriptional activity [8], are diverse and include the Wnt polarity, Wnt-Ca2+, and Wnt-atypical protein kinase C pathways. These pathways have been reported to contribute to developmental processes such as planar cell polarity (PCP), convergent extension movements during gastrulation, neuronal and epithelial cell migration [8-13].
Wnt/Ca2+ signaling, in particular, activates heterotrimeric G proteins that stimulate phospholipase C (PLC). The signaling activation results in intracellular Ca2+ mobilization with activation of Ca2+-dependent effectors that include protein kinase C (PKC), calcium calmodulin mediated kinase II (CAMKII), and calcineurin [14].
Canonical pathway
The canonical pathway is the most studied Wnt signaling pathway as it is involved in a variety of biological processes and integrates signals from other cellular pathways. It controls different processes throughout embryonic development, such as stem cell pluripotency, cell proliferation, differentiation, and cell migration. In adult cells, Wnt signaling contributes to maintain somatic stem cells, regulates cell fate decisions and it is involved in tissue regenerative processes following injury [15].
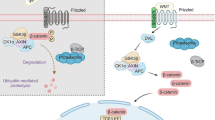
The hallmark of the canonical Wnt pathway is transcriptional activation by β-catenin. The pathway regulates the amount of β-catenin through its degradation or its accumulation and translocation from the adherens junction and cytoplasm into the nucleus. In this way it controls key developmental gene expression programs [7,16,17]. In the absence of Wnt signaling, cytoplasmic β-catenin is constantly degraded by the ubiquitin–proteasome system. This negative regulation involves the multiprotein complex, composed of Axin, adenomatous polyposis coli (APC), casein kinase 1 (CK1), protein phosphatase 2A (PP2A), and glycogen synthase kinase 3β (GSK3β) [18,19]. Axin interacts with the different components of the complex and coordinates sequential phosphorylation of β-catenin. Initially CK1α phosphorylates β-catenin at serine 45 which enables the phosphorylation performed by GSK3β at threonine 41, serine 37, and serine 33 (Figure 1A) [20]. Subsequently, phosphorylation of APC by CK1α and GSK3β leads to an increased affinity between APC and β-catenin triggering a transfer of β-catenin from Axin to APC and to β-Trcp (β-transducin-repeat-containing protein) [21], an E3 ubiquitin ligase subunit that carries out ubiquitination of β-catenin for the proteasome destruction [16,18,22,23].

The canonical Wnt/β-catenin pathway. A) In absence of Wnt signal the destruction complex, formed by the scaffold Axin, APC and GSK3, phosphorilates (P) β-catenin that is then ubiquitinated and degraded via proteasome. In this state the expression of the gene target is repressed. B) In presence of Wnt ligand, the receptor Fzd and the co-receptor LRP5/6 transduce the signal activating Dvl. The destruction complex is inhibited and β-catenin accumulates in the cytoplasm and it translocates into the nucleus. It promotes the target gene expression by binding TCF/LEF and other co-activators. To date several studies identified small molecules (some of these are indicated in the yellow boxes) that can directly inhibit specific components of the Wnt pathway. APC = adenomatous polyposis coli; GSK3 = glycogen synthase kinase; TCF = T cell factor; Fzd = Frizzled receptor; Dvl = Dishevelled.
The Wnt/β-catenin pathway is activated when specific extracellular molecules, Wnt ligands, bind to a receptor complex consisting of a seven-pass transmembrane Frizzled (Fzd) receptor and its co-receptor, low-density lipoprotein receptor related protein 6 (LRP6) or its close relative LRP5. The activated receptors recruit the scaffolding protein Dishevelled (Dvl), which leads to LRP5/6 phosphorylation, mediated by either CK1γ or GSK3β. These events trigger the translocation of Axin to the membrane where it binds to a conserved sequence in the cytoplasmic tail of LRP5/6 [24,25]. Consequently, the APC/Axin/GSK3β complex is destabilized and β-catenin is released allowing it to translocate to the nucleus by a mechanism that is still poorly understood (Figure 1B) [26,27]. In the nucleus, β-catenin binds to the members of the lymphoid enhancer factor T cell (TCF/LEF) DNA-binding transcription factors and induces the expression of downstream targets including c-Myc, cyclin D1, the matrix metalloproteinase MMP-7, the Ets family transcription factor PEA3 and Axin2 [26,28]. In the absence of the Wnt signal, TCF/LEF factors bind DNA at Wnt-responsive genes and interact with other factors (e.g. Groucho, histone deacetylase) to repress gene transcription.
Ligands and the main constituents of the Wnt/β-catenin pathway
The Wnt/β-catenin pathway’s complexity derives from the high number of ligands and receptors involved in Wnt signal transduction that can elicit a variety of intracellular responses [10,29].
Wnt ligands
Wnt ligands comprise a large family of 19 highly conserved cysteine-rich proteins of approximately 350–400 amino acids that contain an N-terminal signal peptide for secretion [16].
Wnt ligands are involved in both the canonical and the non-canonical pathways. Traditionally some ligands (WNT1, WNT3a, and WNT8) have been classified as canonical ligands and others (WNT4, WNT5a, and WNT11) as non-canonical ligands but this classification is now viewed as obsolete. Single Wnt ligands can be involved in multiple intracellular cascades and activate both types of pathways with opposing outcomes. The Wnt outcome depends on the receptor status and on the cellular and microenvironmental context [30,31].
Receptor and co-receptor: Frizzled and LRP
The activation of Wnt/β-catenin signaling requires the cooperation and the aggregation of two types of transmembrane receptors: the Frizzled (Fzd) seven-pass transmembrane G-protein-coupled receptors [32] and the LRP5 and LRP6 [18]. The binding site for Wnt ligands is the extracellular cysteine-rich domain (CRD) that is well conserved between Fzd members [33]. The intracellular C-domain shows sequence diversity among Fzds but a KTxxxW domain is associated with Wnt/β-catenin transduction [31,34,35] and most of Fzd receptors can activate β-catenin signaling [32,36]. In addition to the Fzd-LRP5/6 heterodimerization, Wnt ligands induce LRP5/6 dimerization/oligomerization [26,37] that seems crucial for the canonical pathway activation [16,38]. The ectodomain of LRP5/6 is composed of three LDL repeats (LDLR) and four β-propeller/epidermal growth factor (EGF) repeats (E1-4) that are the binding domain of canonical Wnt ligands and canonical pathway inhibitor Dkk1 [33,38,39]. Chen et al. demonstrated that the receptor complex is maintained in an inactive state when LRP5/6 associates with Fzd. When a Wnt ligand binds to LRP5/6 and Fzd, it is believed to induce a conformational change leading the LRP5/6 dimerization necessary for normal signal transduction [40].
Dvl
Dishevelled (Dvl) proteins are multifunctional intracellular proteins involved in both canonical and non-canonical pathways and have numerous putative binding partners [41]. In mammals there are three isoforms, Dvl-1, 2, and 3, with a modular structure that contains four distinct domains, a DIX, a PDZ and a DEP domain followed by a C-terminal domain (CTD) [42,43]. The DIX and PDZ domains mediate canonical WNT signaling while the PDZ and DEP domains participate in non-canonical pathways. This suggests that Dvl might function as molecular switch regulated by other extracellular signals [41,44]. Indeed Dvl functions are modulated by several phosphorylation sites that are targets of specific kinases and phosphatases [43,45,46].
Axin
Axin is a scaffold protein that acts as a constitutive negative regulator of Wnt signaling by forming a complex with β-catenin, APC, and GSK3β. In particular, this function is carried out by the Axin C-terminal DIX domain (DAX domain) [47-49]. The promotion of rapid and reversible homotypic DAX-DAX polymerization [50] allows the assembly of a dynamic interaction platform that increases the binding affinity for other components such as APC and GSK3β [51]. The Axin-DAX domain can also interact with Dvl-DIX domain forming heterotypic Axin-Dvl interactions: this heteropolymerization switches the Wnt/β-catenin state to being active [52,53]. Axin has another structural domain in its N-terminus (the RGS domain), through which it binds directly to APC [51,54,55]. Axin can be phosphorylated by GSK3 and CK1, and this is believed to increase its association with β-catenin. On the other hand, two serine/threonine phosphatases (PP1 and PP2A) hinder the action of GSK3 and CK1 in the Axin complex reducing the β-catenin degradation. In particular, PP1 dephosphorylates Axin and promotes the disassembly of the Axin complex [16,56].
APC
APC is a tumor suppressor gene located on the long arm of chromosome 5 (5q21). APC has multiple domains that mediate oligomerization as well as binding to a variety of other proteins [57], which have an important role in cell adhesion, signal transduction and transcriptional activation [58]. APC is indispensable for Axin’s activity in assembling the destruction complex [51]. APC may cluster multiple Axin molecules directly, through its multiple Axin-binding sites [55], or indirectly through additional factors (such as CtBP) [59]. Mendoza et al. speculated that APC competes with Dvl for association with Axin, displacing it from Axin protein complex. Wnt signaling may overcome the competition between APC and Dvl for their binding to Axin, allowing simultaneous interaction of all three proteins [37,51,60,61].
APC can be phosphorylated by CK1/GSK3 increasing its affinity for the same β-catenin domain as Axin, suggesting the role of APC in removing the phosphorylated β-catenin molecules from the complex [20,22,62]. Another study suggested that APC protects β-catenin from dephosphorylation by PP2A thereby enhancing β-catenin phosphorylation/degradation [16,56].
CK1
CK1 is a monomeric serine/threonine kinase involved in many different cell functions. There are seven members with high homology: α, β, γ1, γ2, γ3, δ, and ε. Each isoform is involved in different steps of Wnt pathway, with different effects. CKIα is the kinase that first phosphorylates β-catenin at S45, preparing the molecule for the following phosphorylations by GSK3β [46,63,64]. CKIε promotes the activation of Wnt pathway. It phosphorylates Dvl on multiple sites enhancing the binding of GSK3-binding protein/Frat (GBP/Frat) to Dvl [46,65]. CKIε also phosphorylates TCF3 increasing its affinity for β-catenin. CKIγ is anchored on the plasma membrane and it interacts with LRP6 [46,66].
GSK3β
GSK3β is a serine/threonine kinase that is highly conserved from yeast to mammals. In mammals two distinct genes encode two GSK3 isoforms, α (51 kDa) and β (47 kDa), which share 97% amino acid sequence identity within their catalytic domains. The two GSK3 isoforms are ubiquitously expressed and they are involved in a wide variety of essential biological processes such as tissue patterning, glucose metabolism, apoptosis, stem cell homeostasis, and cell cycle regulation [67]. GSK3 has over 40 known direct substrates, and regulates many signaling pathways including the Wnt, MAPK/ERK, BMP, mTOR, and insulin pathways [68,69]. In Wnt signaling, GSK3β is recruited to a multiprotein complex via interaction with Axin, where it phosphorylates β-catenin, marking it for ubiquitination and destruction. Quantitative analysis suggests that the interaction of GSK3β with the Axin enhances phosphorylation of β-catenin by >20000-fold [70]. GSK3 has been proposed to play important roles in human disorders such as bipolar disorder, schizophrenia, Alzheimer disease. It also contributes to neoplastic transformation as it belongs to both the canonical Wnt/β-catenin and the PI3K/Akt signaling systems, two major pathways often dysregulated in cancer [71]. However, to date mutations of GSK3β have not been found in human cancer [72].
CTNNB1 β-catenin
β-catenin has a dual role in the cells: (a) it is a structural protein, stabilizing cell-cell adhesions, which are essential for normal cell physiology and tissue architecture [73,74]; (b) it is the key mediator of canonical Wnt signal transduction from membrane to nucleus, where it operates as a transcriptional co-activator of the T cell factor (TCF) family of DNA-binding proteins [75]. β-catenin provides a direct connection between extracellular signals, gene transcription and cell cycle control [29,73].
β-catenin protein has three domains: the N-terminal domain, the armadillo domain consisting of 12 armadillo repeats (residues 141–664), and a C-terminal domain. The positively charged armadillo (Arm) repeat is the binding site for most β-catenin binding partners [76,77]. Local charge alterations of β-catenin through phosphorylation at multiple sites have the ability to regulate its affinity to specific protein partners. Phosphorylation at the C-terminal domain decreases the binding of β-catenin to the cadherin adhesion complex, while the N-terminal domain is the site of GSK3 and CK1 phosphorylation which is recognized by the β-TrCP ubiquitin ligase for the β-catenin degradation [78,79].
Endogenous inhibitors of Wnt
Wnt/β-catenin signaling is endogenously regulated by secreted proteins that antagonize the Wnt ligands and act at the cell surface level in order to inhibit the pathway [80]. Among these, there are secreted frizzled-related proteins (sFRP) and Wnt inhibitor proteins (WIF) that inhibit the interaction between Wnt and its receptors. Another inhibitor, the Dickkopf related protein 1 (DKK-1), is a ligand for the Wnt coreceptors LRP5/6 [81]. DKK-1 antagonizes LRP6 function by disrupting Fzd-LRP6 complex or by interacting with LRP6 and consequently promoting its internalization and degradation (Figure 1A) [82].
Role of the Wnt pathway in human pathology
Deregulations of the Wnt pathway are linked to several human diseases comprising various types of cancer (including skin, breast and colon cancers), skeletal defects, birth defect disorders (including neural tube defects), fibrotic diseases, metabolic diseases, neurodegenerative disorders and others [7,83]. Several causes can lead to alterations in the Wnt pathway including germline and somatic mutations, epigenetic modifications as well as microenvironmental factors.
Wnt pathway and genetic disorders
The Wnt pathway has been extensively investigated for its involvement in many types of cancer [28]. Several studies with experimental models demonstrated that a high level of β-catenin activity is required for tumor initiation [70]. In particular, colorectal cancer, desmoid tumor, gastric cancer, melanoma, hepatocellular, prostate, thyroid, ovarian, endometrial cancer, and some subsets of breast cancers harbour β-catenin-stabilizing mutations, including germline APC gene and somatic CTNNB1 gene mutations [30,72,75]. Genetic alterations of Axin2 has been described in adrenocortical carcinoma [84], hepatocellular carcinoma and it may predispose to colorectal cancer [80,85]. Patients with distinct types of hereditary high bone mass diseases were found to carry mutations in the LRP5 extracellular domain, while mutations in LRP6 are linked to hereditary disorders as osteoporosis, coronary artery disease, and metabolic syndrome [80]. Mutations in LRP5 and TCF7L2 genes may lead to the development of obesity and mellitus diabetes [86,87].
APC gene mutations
The association between colon cancer and the aberrant regulation of the Wnt pathway has been known since the identification of alterations of chromosome 5q as an early event in the carcinogenic process for hereditary colon tumors (Familial Adenomatous Polyposis, FAP), and the discovery, through different linkage studies, of the APC gene at this chromosomal site [88,89].
FAP is a colon cancer predisposition syndrome, which is inherited in an autosomal dominant manner. Clinical diagnosis of FAP can be made when more than 100 adenomatous polyps are identified in the colorectum. FAP patients present not only colorectal adenomas but also various extracolonic manifestations, including desmoid tumors, osteomas, dental abnormalities, congenital hypertrophy of the retinal pigment epithelium, lipomas, epidermoid cysts and upper gastrointestinal polyps.
To date, more than 300 different APC gene mutations are recognized as the cause of FAP. Most of these mutations (insertions, deletions, nonsense mutations, etc.) cause a truncated or inactive protein [58]. APC mutations have been subsequently found in ~80% of sporadic colorectal tumors, confirming that APC acts as a central gatekeeper protein in colorectal tumorigenesis [90].
Inherited or somatic mutations that inactivate or destroy APC function prevent effective degradation of β-catenin, promoting the aberrant activation of canonical Wnt signaling. This leads to the development of non-invasive colonic adenomas (polyps) because β-catenin nuclear accumulation causes the overexpression of growth-promoting genes [91]. The same outcome can arise through mutations in CTNNB1 and AXIN2, though these are significantly less frequent than mutations in APC [92].
Epigenetic modifications affecting the Wnt pathway
In addition to the genetic mutations, epigenetic modifications can contribute to aberrant activation of the canonical Wnt pathway. This can occur at various levels and determines the silencing or promoting of specific genes. In particular, aberrant methylation of CpG islands within gene promoter regions represents one of the most studied mechanisms of gene silencing and it is associated with selective transcriptional inactivation.
Many evidences indicate, for example, that almost complete loss of SFRPs at the protein levels are frequently correlated with gene promoter hypermethylation in several pathologies such as colon carcinomas, hepatocarcinomas [93], prostate cancer, human brain cancers [94], non-small cell lung cancer [95], esophageal carcinoma [96], myeloproliferative neoplasms.
A loss of SFRP expression, through epigenetic silencing, contributes to the constitutive activation of autocrine Wnt signaling affecting cell proliferation and potentially enhancing the cell growth and promoting malignant transformation and cancer cell survival [1,2,97,98].
Wnt pathway’s interaction with the microenvironment
Regulators of the microenvironment, such as the extracellular matrix, growth factors and inflammatory factors, are associated with the aberrant activation of Wnt pathway and the promotion of several diseases.
Inflammation and Wnt pathway signaling
Inflammation is a critical defense mechanism against various harmful stimuli, although aberrant regulation may lead to diseases. The inflammation process, caused by injury, leads to wound healing, tissue repair and regeneration. Damaged epithelial and endothelial cells release inflammatory factors, growth factors, cytokines, and chemokines, which subsequently initiate an influx of neutrophils and monocytes to the site of the damaged tissue. Macrophages secrete platelet-derived growth factor (PDGF), connective tissue growth factor (CTGF) and transforming growth factor-β (TGF-β). They also activate and convert fibroblasts into myofibroblasts, which are engaged in extracellular matrix (ECM) deposition and scar formation [99,100]. Cytokines activate gene transcription regulators that are involved in stem cell renewal and proliferation, critical for tissue repair [100-103]. In case of repetitive injuries or unresolved damage, the inflammatory process can lead to aberrant fibroblast activation and excessive ECM accumulation with subsequent tissue fibrosis that can evolve into fibrotic disease and potential tumor initiation [100,101,104].
GSK3 has a crucial role in inflammation because it promotes pro-inflammatory cytokine production (IL-6, IL-1β and TNF-α) and cell migration [71,105,106]. Furthermore, two major pro-inflammatory cytokines, IFNγ and TNFα, are key regulators of β-catenin signaling and the most highly induced mediators in the inflamed tissue [107]. Thus there is a lasting involvement of Wnt/β-catenin signaling during the inflammation process that is associated with pathogenic disorder.
Wnt pathway and fibro-proliferative diseases
The development and progression of several fibrotic diseases is the outcome of the Wnt pathway cross-talk with other signaling pathways and pro-inflammatory mediators [108].
In general, aberrant Wnt/β-catenin signaling activation drives fibrogenesis through interaction with profibrotic growth factors, epithelial cell transformation, myofibroblasts activation and proliferation [109]. Mutant mice models demonstrate the involvement of β-catenin signaling in fibroproliferative diseases [110,111]. Furthermore, in fibrotic diseases, Wnts and positive regulators of β-catenin are upregulated, whereas inhibitors of Wnt/β-catenin signaling are downregulated [110].
The cross-talk between Wnt/β-catenin and TGF-β pathways has been demonstrated in several fibroproliferative disorders such as Dupuytren’s disease and pulmonary fibrosis [112,113]. TGF-β regulates the fibroblast activation to myofibroblast [81,108]. Wei et al. showed that Wnt3a activates the TGF-β cascade inducing the expression of pro-fibrotic genes [81,108]. On the other hand, TGF-β signaling seems to up-regulate Wnt/β-catenin pathway by decreasing the expression of Dkk-1, which in turn, inhibits the canonical Wnt pathway [81].
Desmoid-type fibromatosis: A pathology arising from Wnt pathway genetic alteration and microenvironmental factors
Desmoid-type fibromatosis (DFs) can be an example of pathology arising from direct Wnt/β-catenin signaling alteration (Wnt mediator mutations) as well as indirect Wnt deregulation by involvement of the microenvironment. DF is a rare myofibroblastic neoplasm arising from a defect in connective tissue regulation, the neoplasia is unable to metastasize but it shows marked local aggression and a high recurrence rate. Some DFs are consequence of local trauma including pregnancies and surgical treatments [114,115]; repeated injuries also might increase the risk of DF recurrence.
Genetic cause of desmoid-type fibromatosis: mutations of CTNNB1 gene
Desmoid-type fibromatosis might be one of the manifestations of the APC gene linked FAP but they are generally sporadic tumors. A range from 50% to 87% of sporadic DFs are characterized by mutations in codons 41 and 45 of exon 3 (p.Thr41Ala, p.Ser45Phe, and p.Ser45Pro) of the gene encoding β-catenin, CTNNB1. These codons are the serine and threonine phosphorylation sites required for β-catenin degradation [116,117]. Therefore, these mutations make phosphorylation impossible and promote β-catenin nuclear translocation [118]. A high level of nuclear β-catenin staining is the conventional diagnostic marker for DFs. Nuclear β-catenin is detected in almost 90% of the desmoid cells (Figure 2A). However, the abnormal expression of β-catenin is independent of CTNNB1 mutations, suggesting that other factors might be involved in the alteration of the Wnt/β-catenin pathway in DFs. Intriguingly, in all DF cells, we have also noticed a very marked increase in nuclear GSK3β (95%) associated to β-catenin, suggesting that other changes involving the multiprotein complex are involved with the disease (Figure 2B) [118]. In addition, Caspi et al. demonstrated that GSK3β may have a nuclear function that impairs the Wnt pathway by a mechanism that does not involve phosphorylation and degradation of β-catenin [119,120]. These results support the potential significance of nuclear GSK3β as an additional marker for DF cells [118].

Nuclear localization of GSK-3β and β-catenin in desmoid-type fibromatosis (DF) cells. A) DF cells and control cells (ctr) were immunostained with anti-β-catenin (red). The nucleus was stained with DAPI (blue). The pictures show the nuclear localization of β-catenin in DF cells, and cytoplasmic staining in control cells. B) DF cells were immunostained with anti-β-catenin (green) and GSK-3β (red) antibodies. The nucleus was stained with DAPI. The merged picture shows colocalization of β-catenin and GSK-3β.
Microenvironmental origin of desmoid-type fibromatosis
Nuclear accumulation of β-catenin in DFs can be also caused by microenvironmental factors such as inflammation, growth factors or hormones. Immunohistochemistry studies demonstrated that EGF, TGF-β, TNF-α, VEGF, phosphorylated SMAD2/3, COX2, and androgen receptor were significantly increased in desmoid tumors compared with healing scar tissue and quiescent fibrous tissue [121-124].
TGF-β is a modulator of β-catenin levels. Cultured fibroblasts, stimulated with TGF-β, induced nuclear accumulation of β-catenin and increased the activity of TCF/LEF reporter and transcription of the target gene AXIN2 [81,108]. Expression of TGF-β-related cytokines has also been described in desmoid tumors [121,123-125].
Human DF samples also showed expression of the PDGFα and PDGFRα, metalloproteinases, ADAM12 and MMP2, and midkine, heparin-binding growth factor [126,127]. Expression of progesteron receptors has been reported in DFs samples, while they were negative for the estrogen receptor alpha [128,129].
Perspectives of therapeutic approaches
Wnt pathway inhibitors
As the canonical Wnt signaling is one of the central profibrotic signaling pathways [130] its inhibition on different levels (from ligand secretion to intracellular mediators) might be an effective antifibrotic treatment. Overexpression of the endogenous inhibitor Dkk-1 strongly ameliorated fibrosis in in vitro models mimicking early or later stages of human disease [108]. Thus it may be an attractive target for treating fibrosis, microvascular inflammation, tubule injury, and microvascular rarefaction [131]. However, the most effective therapy would be targeted the downstream complex in the pathway by using TCF/β-catenin antagonist that inhibits protein-protein interaction between TCF and β-catenin. Beyer and collaborators evaluated the antifibrotic effects of two small molecules, PKF118-310 and ICG-001, in the inflammatory model of bleomycin-induced dermal fibrosis. While PKF118-310 inhibits the β-catenin/TCF interaction, ICG-001 interferes with the recruitment of co-activators to β-catenin. The treatment with PKF118-310 and ICG-001 effectively inhibited canonical Wnt signaling reducing mRNA expression of Axin-2 (Figure 1B) [130]. These compounds prevent and reverse inflammation-driven fibrosis and reduce TGF-β dependent fibrosis.
Another mechanism for decreasing canonical Wnt signalling is to target the PDZ domain of DVL. Three compounds (NSC 668036, FJ9 and 3289–8625) have been identified to in vitro inhibit the Frizzled receptor-PDZ domain interaction.
Furthermore, the level of Axin in the β-catenin destruction complex is controlled by tankyrases, members of the PARP-family of poly-ADP-ribosylation enzymes. Small molecules, inhibiting the tankyrase 1 and tankyrase 2 enzymes, stabilize the level of Axin and promote the phosphorylation-dependent degradation of β-catenin by increasing the activity of the destruction complex [132]. Among these molecules Wang and collaborators demonstrated that XAV939 significantly inhibited the activation of Wnt/β-catenin signalling and attenuated bleomycin-induced lung fibrosis in mice [133]. The reduction of Wnt/β-catenin signaling, by the tankyrase inhibitors G007-LK and G244-LM, has been also demonstrated in APC mutant colorectal cancer (CRC) cell lines [134]. However, the clinical use of these inhibitors may be limited by the intestinal toxicity in APC-mutant CRC models and local or systemic toxicity in the fibrotic tissue of systemic sclerosis [134]. Intriguing, the pharmacological manipulation of Wnt pathway, using GSK3β inhibitors (lithium chloride, SB216763) (Figure 1A), is a promising therapeutic approach for several pathologies such as diabetes, stroke, mood disorders, inflammation, and Alzheimer’s disease [135].
Moreover, as GSK3 is a vital factor in inflammatory processes, inhibitors of GSK3 provide strong anti-inflammatory protection. GSK3 inhibitors were reported to reduce the inflammatory response in induced colitis in rats, as well as in arthritis and peritonitis in mice highlighting the potential therapeutic treatments in pathological conditions associated to inflammation [71,136,137].
Therapeutic treatments described in desmoid-type fibromatosis
DF treatment is complicated by its recurrence, invasiveness, and persistence. Due to the heterogeneity of the desmoid-type fibromatosis and to the unpredictable clinical course, at the moment, the treatment is given on a case-by-case multimodal basis [138-140]. For this reason and for the absence of metastatic potential the “wait and see approach” is preferred when the tumors are asymptomatic and not located in area that could lead to functional limitations [141]. On the other hand, when the tumour mass causes discomfort, affects the function of involved organs or causes severe cosmetic damage, surgery is the preferred option, in association with radiotherapy and/or chemotherapy [142-144]. When the tumors are unresectable, radiotherapy is recommended [145,146]. For abdominal tumors, systemic therapy with non steroidal anti-inflammatory drugs, hormonal or biological agents, and cytotoxic drugs, is suggested. Different drugs have been used in clinics with different outcomes including Tamoxifen, Interferon-α, Doxorubicin, Imatinib and Sorafenib [146-150]. In particular, Imatinib mesylate has been reported to inhibit receptor tyrosine kinases, including PDGFR-α and PDGFR-β, as well as c-kit [151]. As desmoid tumor cells produce high levels of TGF-β, Toremifene which is an antiestrogen that inhibits collagen and TGF-β synthesis, has been used for in vitro desmoid cells. The results showed the reduction of receptor number only in desmoid cells, suggesting that Toremifene may reduce TGF-β's affinity for its receptors [121,152]. Toremifene also modifies the ECM components that regulate cytokine activity and cell migration.
An experimental animal model demonstrated that Apc(+)/Apc(1638 N) mice treated with Triparanol, an inhibitor of Hedgehog (Hh) signaling, develop few and smaller desmoid tumors compared with the untreated mice [153]. These data provide functional evidence that Hh pathway, associated with aberrant Wnt pathway, plays a key role in the maintenance of normal cells as the modulation of this pathway influences desmoid tumor behaviour. It also suggests Hh blockade as a therapeutic approach for this tumor type [153]. Hyperthermic isolated limb perfusion with TNF-α and Melphalan resulted to be an effective treatment in desmoid tumor recurrence of the limb or where resection threatens loss of function [154-156].
Conclusions
The Wnt/β-catenin pathway is a great example of heavily context dependent cellular pathways with several ligands, receptors, transmitters and modulators. In general the interactions of the Wnt/β-catenin pathway with the other cellular processes clearly state its importance for the cell and the entire organism.
The most direct therapeutic approach against the deregulation of the Wnt/β-catenin pathway is to target the components of the pathways themselves or their closest interactors.
The dependence of the Wnt/β-catenin pathway on its microenvironment can be exploited as a potential target for therapeutic approaches in particular the host’s response to pathological cells including inflammation and the various growth factors produced in the attempt to “heal” the organism. This approach could also lead to a faster individuation of a valid treatment as several compounds are already commercialized, even if they are developed for completely unrelated diseases.
In synthesis, we need to continue studying on two fronts in order to find effective treatments for Wnt/β-catenin pathway related pathologies: the Wnt/β-catenin pathway itself and its role in the network comprising other pathways associated to the microenvironment.
Consent
An informed written consent was obtained from the persons whose cell culture images were included in this review.
Abbreviations
- ADAM:
-
Disintegrin and metalloproteinase domain-containing protein
- APC:
-
Adenomatous polyposis coli
- β-Trcp:
-
β-transducin-repeat-containing protein
- BMP:
-
Bone morphogenetic protein
- CAMKII:
-
Calmodulin mediated kinase II
- CK1:
-
Casein kinase 1
- COX:
-
Cytochrome c oxidase
- CtBP:
-
C-terminal-binding protein
- CTD:
-
C-terminal domain
- CTGF:
-
Connective tissue growth factor
- CTNNB1:
-
Catenin beta-1
- DF:
-
Desmoid-type fibromatosis
- DKK-1:
-
Dickkopf related protein 1
- Dvl:
-
Dishevelled
- ECM:
-
Extracellular matrix
- EGF:
-
Epidermal growth factor
- FAP:
-
Familial adenomatous polyposis
- Fzd:
-
Frizzled receptor
- GBP/Frat:
-
GSK3 binding protein/Frat
- GSK3:
-
Glycogen synthase kinase 3
- Hh:
-
Hedgehog
- IFN-γ:
-
Interferon gamma
- IL:
-
Interleukin
- LDRL:
-
Low-density lipoprotein repeats
- LRP:
-
Low-density lipoprotein receptor related protein
- MAPK/ERK:
-
Mitogen-activated protein kinase/extracellular regulated MAP kinase
- MMP:
-
Matrix metalloproteinase
- mTOR:
-
Mammalian target of rapamycin
- PCP:
-
Planar cell polarity
- PDGF:
-
Platelet-derived growth factor
- PDGFR:
-
Platelet-derived growth factor receptor
- PI3K/Akt:
-
Phosphotidylinositol 3 kinase/serine/threonine-protein kinase
- PKC:
-
Protein kinase C
- PLC:
-
Phospholipase C
- PP1:
-
Protein phosphatase 1
- PP2A:
-
Protein phosphatase 2A
- sFRP:
-
Secreted frizzled related-protein
- SMAD:
-
Small mothers against decapentaplegic
- TCF/LEF:
-
T cell factor/lymphoid enhancer factor
- TGF-β:
-
Transforming growth factor β
- TNF-α:
-
Tumor necrosis factor-α
- VEGF:
-
Vascular endothelial growth factor
- WIF:
-
Wnt inhibitor protein
References
Bafico A, Liu G, Goldin L, Harris V, Aaronson SA. An autocrine mechanism for constitutive Wnt pathway activation in human cancer cells. Cancer Cell. 2004;6:497–506.
Akiri G, Cherian MM, Vijayakumar S, Liu G, Bafico A, Aaronson SA. Wnt pathway aberrations including autocrine Wnt activation occur at high frequency in human non-small-cell lung carcinoma. Oncogene. 2009;28:2163–72.
Eisenmann DM. Wnt signaling. WormBook. 2005;25:1–17.
Peifer M, Polakis P. Wnt signaling in oncogenesis and embryogenesis–a look outside the nucleus. Science. 2000;287:1606–9.
Hobmayer B, Rentzsch F, Kuhn K, Happel CM, von Laue CC, Snyder P, et al. WNT signalling molecules act in axis formation in the diploblastic metazoan Hydra. Nature. 2000;407:186–9.
Wodarz A, Nusse R. Mechanisms of Wnt signaling in development. Annu Rev Cell Dev Biol. 1998;14:59–88.
Komiya Y, Habas R. Wnt signal transduction pathways. Organogenesis. 2008;4:68–75.
Semenov MV, Habas R, Macdonald BT, He X. SnapShot: noncanonical Wnt signaling pathways. Cell. 2007;131:1378.
Simons M, Mlodzik M. Planar cell polarity signaling: from fly development to human disease. Annu Rev Genet. 2008;42:517–40.
Kikuchi A, Yamamoto H, Sato A. Selective activation mechanisms of Wnt signaling pathways. Trends Cell Biol. 2009;19:119–29.
Castanon I, Abrami L, Holtzer L, Heisenberg CP, van der Goot FG, González-Gaitán M. Anthrax toxin receptor 2a controls mitotic spindle positioning. Nat Cell Biol. 2013;15:28–39.
Wu J, Roman AC, Carvajal Gonzalez JM, Mlodzik M. Wg and Wnt4 provide long-range directional input to planar cell polarity orientation in Drosophila. Nat Cell Biol. 2013;15:1045–55.
Zallen JA. Planar polarity and tissue morphogenesis. Cell. 2007;129:1051–63.
van Amerongen R. Alternative Wnt pathways and receptors. Cold Spring Harb Perspect Biol. 2012;1:4(10).
Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013;13:11–26.
MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26.
Jamieson C, Sharma M, Henderson BR. Regulation of β-catenin nuclear dynamics by GSK-3β involves a LEF-1 positive feedback loop. Traffic. 2011;12:983–99.
He X, Semenov M, Tamai K, Zeng X. LDL receptor-related proteins 5 and 6 in Wnt/beta-catenin signaling: arrows point the way. Development. 2004;131:1663–77.
Gordon MD, Nusse R. Wnt signaling: multiple pathways, multiple receptors, and multiple transcription factors. J Biol Chem. 2006;281:22429–33.
Kimelman D, Xu W. beta-catenin destruction complex: insights and questions from a structural perspective. Oncogene. 2006;25:7482–91.
Huber AH, Weis WI. The structure of the -catenin/E-cadherin complex and the molecular basis of diverse ligand recognition by -catenin. Cell. 2001;105:391–402.
Xing Y, Clements WK, Kimelman D, Xu W. Crystal structure of a beta-catenin/axin complex suggests a mechanism for the beta-catenin destruction complex. Genes Dev. 2003;17:2753–64.
Hart M, Concordet JP, Lassot I, Albert I, Del Los SR, Durand H, et al. The F-box protein beta-TrCP associates with phosphorylated beta-catenin and regulates its activity in the cell. Curr Biol. 1999;9:207–10.
Mao J, Wang J, Liu B, Pan W, Farr 3rd GH, Flynn C, et al. Low-density lipoprotein receptor-related protein-5 binds to Axin and regulates the canonical Wnt signaling pathway. Mol Cell. 2001;7:801–9.
Zeng X, Huang H, Tamai K, Zhang X, Harada Y, Yokota C, et al. Initiation of Wnt signaling: control of Wnt coreceptor Lrp6 phosphorylation/activation via frizzled, dishevelled and axin functions. Development. 2008;135:367–75.
Cong F, Varmus H. Nuclear-cytoplasmic shuttling of Axin regulates subcellular localization of beta-catenin. Proc Natl Acad Sci U S A. 2004;101:2882–7.
Schwarz-Romond T, Metcalfe C, Bienz M. Dynamic recruitment of axin by Dishevelled protein assemblies. J Cell Sci. 2007;120:2402–12.
Reya T, Clevers H. Wnt signaling in stem cells and cancer. Nature. 2005;434:843–50.
van Amerongen R, Nusse R. Towards an integrated view of Wnt signalling in development. Development. 2009;136:3205–14.
Najdi R, Holcombe RF, Waterman ML. Wnt signaling and colon carcinogenesis: beyond APC. J Carcinog. 2011;17:10–5.
Ring L, Neth P, Weber C, Steffens S, Faussner A. β-Catenin-dependent pathway activation by both promiscuous "canonical" WNT3a-, and specific "non canonical"WNT4- and WNT5a FZD receptor combinations with strong differences in LRP5 and LRP6 dependency. Cell Signal. 2014;26:260–7.
Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810.
Bhanot P, Brink M, Samos CH, Hsieh JC, Wang Y, Macke JP, et al. A new member of the frizzled family from Drosophila functions as a Wingless receptor. Nature. 1996;18:225–30.
Umbhauer M, Djiane A, Goisset C, Penzo-Méndez A, Riou JF, Boucaut JC, et al. The C-terminal cytoplasmic Lys-thr-X-X-X-Trp motif in frizzled receptors mediates Wnt/beta-catenin signalling. EMBO J. 2000;19:4944–54.
Punchihewa C, Ferreira AM, Cassell R, Rodrigues P, Fujii N. Sequence requirement and subtype specificity in the high-affinity interaction between human frizzled and dishevelled proteins. Protein Sci. 2009;18:994–1002.
Binnerts ME, Kim KA, Bright JM, Patel SM, Tran K, Zhou M, et al. R-Spondin1 regulates Wnt signaling by inhibiting internalization of LRP6. Proc Natl Acad Sci U S A. 2007;104:14700–5.
Bilic J, Huang YL, Davidson G, Zimmermann T, Cruciat CM, Bienz M, et al. Wnt induces LRP6 signalosomes and promotes dishevelled-dependent LRP6 phosphorylation. Science. 2007;316:1619–22.
Angers S, Moon RT. Proximal events in Wnt signal transduction. Nat Rev Mol Cell Biol. 2009;10:468–77.
Chen S, Bubeck D, MacDonald BT, Liang WX, Mao JH, Malinauskas T, et al. Structural and functional studies of LRP6 ectodomain reveal a platform for Wnt signaling. Dev Cell. 2011;21:848–61.
Chen J, Yan H, Ren DN, Yin Y, Li Z, He Q, et al. LRP6 dimerization through its LDLR domain is required for robust canonical Wnt pathway activation. Cell Signal. 2014;26:1068–74.
Wallingford JB, Habas R. The developmental biology of Dishevelled: an enigmatic protein governing cell fate and cell polarity. Development. 2005;132:4421–36.
Seifert JR, Mlodzik M. Frizzled/PCP signalling: a conserved mechanism regulating cell polarity and directed motility. Nat Rev Genet. 2007;8:126–38.
González-Sancho JM, Greer YE, Abrahams CL, Takigawa Y, Baljinnyam B, Lee KH, et al. Functional consequences of Wnt-induced dishevelled 2 phosphorylation in canonical and noncanonical Wnt signaling. J Biol Chem. 2013;288:9428–37.
Grumolato L, Liu G, Mong P, Mudbhary R, Biswas R, Arroyave R, et al. Canonical and noncanonical Wnts use a common mechanism to activate completely unrelated coreceptors. Genes Dev. 2010;24:2517–30.
Yanfeng WA, Berhane H, Mola M, Singh J, Jenny A, Mlodzik M. Functional dissection of phosphorylation of Disheveled in Drosophila. Dev Biol. 2011;360:132–42.
Price MA. CKI, there’s more than one: casein kinase I family members in Wnt and Hedgehog signaling. Genes Dev. 2006;20:399–410.
Sakanaka C, Williams LT. Functional domains of axin: importance of the C terminus as an oligomerization domain. J Biol Chem. 1999;274:14 090–3.
Fiedler M, Mendoza-Topaz C, Rutherford TJ, Mieszczanek J, Bienz M. Dishevelled interacts with the DIX domain polymerisation interface of Axin to interfere with its function in downregulating b-catenin. Proc Natl Acad Sci U S A. 2011;108:1937–42.
Faux MC, Coates JL, Catimel B, Cody S, Clayton AH, Layton MJ, et al. Recruitment of Adenomatous polyposis coli and b-catenin to axin-puncta. Oncogene. 2008;27:5808–20.
Schwarz-Romond T, Merrifield C, Nichols BJ, Bienz M. The Wnt signalling effector Dishevelled forms dynamic protein assemblies rather than stable associations with cytoplasmic vesicles. J Cell Sci. 2005;118:5269–77.
Mendoza-Topaz C, Mieszczanek J, Bienz M. The Adenomatous polyposis coli tumour suppressor is essential for Axin complex assembly and function and opposes Axin's interaction with Dishevelled. Open Biol. 2011;3:110013.
Luo W, Zou H, Jin L, Lin S, Li Q, Ye Z, et al. Axin contains three separable domains that confer intramolecular, homodimeric, and heterodimeric interactions involved in distinct functions. J Biolo Chem. 2005;280:5054–60.
Schwarz-Romond T, Fiedler M, Shibata N, Butler PJ, Kikuchi A, Higuchi Y, et al. The DIX domain of Dishevelled confers Wnt signaling by dynamic polymerization. Nat Struct Mol Biol. 2007;14:484–92.
Behrens J, Jerchow BA, Wurtele M, Grimm J, Asbrand C, Wirtz R, et al. Functional interaction of an axin homolog, conductin, with β-catenin, APC, and GSK3b. Science. 1998;280:596–9.
Spink KE, Polakis P, Weis WI. Structural basis of the Axin–Adenomatous polyposis coli interaction. EMBO J. 2000;19:2270–9.
Su Y, Fu C, Ishikawa S, Stella A, Kojima M, Shitoh K, et al. APC is essential for targeting phosphorylated beta-catenin to the SCF(beta-TrCP) ubiquitin ligase. Mol Cell. 2008;32:652–61.
Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66:589–600.
Half E, Bercovich D, Rozen P. Familial adenomatous polyposis. Orphanet J Rare Dis. 2009;4:22.
Schneikert J, Brauburger K, Behrens J. APC mutations in colorectal tumours from FAP patients are selected for CtBP-mediated oligomerization of truncated APC. Hum Mol Genet. 2011;20:3554–64.
Katsanis N, Fisher EM. A novel C-terminal binding protein (CTBP2) is closely related to CTBP1, an adenovirus E1A-binding protein, and maps to human chromosome 21q21.3. Genomics. 1998;47:294–9.
Cliffe A, Hamada F, Bienz M. A role of Dishevelled in relocating Axin to the plasma membrane during Wingless signaling. Curr Biol. 2003;13:960–6.
Valvezan AJ, Zhang F, Diehl JA, Klein PS. Adenomatous polyposis coli (APC) regulates multiple signaling pathways by enhancing glycogen synthase kinase-3 (GSK-3) activity. J Biol Chem. 2012;287:3823–32.
Amit S, Hatzubai A, Birman Y, Andersen JS, Ben-Shushan E, Mann M, et al. Axin-mediated CKI phosphorylation of β-catenin at Ser 45: A molecular switch for the Wnt pathway. Genes Dev. 2002;16:1066–76.
Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, et al. Control of β-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002;108:837–47.
Hino S, Michiue T, Asashima M, Kikuchi A. Casein kinase Iε enhances the binding of Dvl-1 to Frat-1 and is essential for Wnt-3a-induced accumulation of β-catenin. J Biol Chem. 2003;278:14066–73.
Davidson G, Wu W, Shen J, Bilic J, Fenger U, Stannek P, et al. Casein kinase 1γ couples Wnt receptor activation to cytoplasmic signal transduction. Nature. 2005;438:867–72.
Fodde R, Brabletz T. Wnt/beta-catenin signaling in cancer stemness and malignant behavior. Curr Opin Cell Biol. 2007;19:150–8.
Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8:387–98.
Brown RL, Reinke LM, Damerow MS, Perez D, Chodosh LA, Yang J, et al. CD44 splice isoform switching in human and mouse epithelium is essential for epithelial-mesenchymal transition and breast cancer progression. J Clin Invest. 2011;121:1064–74.
Dajani R, Fraser E, Roe SM, Yeo M, Good VM, Thompson V, et al. Structural basis for recruitment of glycogen synthase kinase 3beta to the axin-APC scaffold complex. EMBO J. 2003;22:494–501.
Jope RS, Yuskaitis CJ, Beurel E. Glycogen synthase kinase-3 (GSK3): inflammation, diseases, and therapeutics. Neurochem Res. 2007;32:577–95.
Rosenbluh J, Wang X, Hahn WC. Genomic insights into WNT/β-catenin signaling. Trends Pharmacol Sci. 2014;35:103–9.
Beyer C, Schramm A, Akhmetshina A, Dees C, Kireva T, Gelse K, et al. β-catenin is a central mediator of pro-fibrotic Wnt signaling in systemic sclerosis. Ann Rheum Dis. 2012;71:761–7.
Conacci-Sorrell M, Zhurinsky J, Ben-Ze’ev A. The cadherin-catenin adhesion system in signaling and cancer. J Clin Invest. 2002;109:987–91.
Jamieson C, Sharma M, Henderson BR. Targeting the β-catenin nuclear transport pathway in cancer. Semin Cancer Biol. 2014;27:20–9.
Huber AH, Nelson WJ, Weis WI. Three-dimensional structure of the armadillo repeat region of beta-catenin. Cell. 1997;90:871–82.
Xu W, Kimelman D. Mechanistic insights from structural studies of beta-catenin and its binding partners. J Cell Sci. 2007;120:3337–44.
Jiang J, Struhl G. Regulation of the Hedgehog and Wingless signalling pathways by the F-box/WD40-repeat protein Slimb. Nature. 1998;391:493–6.
Daugherty RL, Gottardi CJ. Phospho-regulation of Beta-catenin adhesion and signaling functions. Physiology. 2007;22:303–9.
Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell. 2012;149:1192–205.
Akhmetshina A, Palumbo K, Dees C, Bergmann C, Venalis P, Zerr P, et al. Activation of canonical Wnt signalling is required for TGF-β-mediated fibrosis. Nat Commun. 2012;13:3–735.
Semënov MV, Zhang X, He X. DKK1 antagonizes Wnt signaling without promotion of LRP6 internalization and degradation. J Biol Chem. 2008;283:21427–32.
Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–80.
Guimier A, Ragazzon B, Assié G, Tissier F, Dousset B, Bertherat J, et al. AXIN genetic analysis in adrenocortical carcinomas updated. J Endocrinol Invest. 2013;36:1000–3.
Lammi L, Arte S, Somer M, Jarvinen H, Lahermo P, Thesleff I, et al. Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am J Hum Genet. 2004;74:1043–50.
Kahn M. Can we safely target the WNT pathway? Nat Rev Drug Discov. 2014;13:513–32.
Jin T. The WNT signalling pathway and diabetes mellitus. Diabetologia. 2008;51:1771–80.
Nishisho I, Nakamura Y, Miyoshi Y, Miki Y, Ando H, Horii A, et al. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science. 1991;253:665–9.
Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–70.
Nieuwenhuis MH, Mathus-Vliegen LM, Slors FJ, Griffioen G, Nagengast FM, Schouten WR, et al. Genotype-phenotype correlations as a guide in the management of familial adenomatous polyposis. Clin Gastroenterol Hepatol. 2007;5:374–8.
Polakis P. The oncogenic activation of beta-catenin. Curr Opin Genet Dev. 1999;9:15–21.
Liu W, Dong X, Mai M, Seelan RS, Taniguchi K, Krishnadath KK, et al. Mutations in AXIN2 cause colorectal cancer with defective mismatch repair by activating beta-catenin/TCF signalling. Nat Genet. 2000;26:146–7.
Xie Q, Chen L, Shan X, Shan X, Tang J, Zhou F, et al. Epigenetic silencing of SFRP1 and SFRP5 by hepatitis B virus X protein enhances hepatoma cell tumorigenicity through Wnt signalling pathway. Int J Cancer. 2014;135:635–46.
Schiefer L, Visweswaran M, Perumal V, Arfuso F, Groth D, Newsholme P, et al. Epigenetic regulation of the secreted frizzled-related protein family in human glioblastoma multiforme. Cancer Gene Ther. 2014;21:297–303.
Stewart DJ, Chang DW, Ye Y, Spitz M, Lu C, Shu X, et al. Wnt signalling pathway pharmacogenetics in non-small cell lung cancer. Pharmacogenomics J. 2014;14:509–22.
Saito T, Mitomi H, Imamhasan A, Hayashi T, Mitani K, Takahashi M, et al. Downregulation of sFRP-2 by epigenetic silencing activates the β-catenin/Wnt signaling pathway in esophageal basaloid squamous cell carcinoma. Virchows Arch. 2014;464:135–43.
Schlange T, Matsuda Y, Lienhard S, Huber A, Hynes NE. Autocrine WNT signalling contributes to breast cancer cell proliferation via the canonical WNT pathway and EGFR transactivation. Breast Cancer Res. 2007;9:R63.
Vijayakumar S, Liu G, Rus IA, Yao S, Chen Y, Akiri G, et al. High-frequency canonical Wnt activation in multiple sarcoma subtypes drives proliferation through a TCF/β-catenin target gene, CDC25A. Cancer Cell. 2011;19:601–12.
Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210.
Kuraishy A, Karin M, Grivennikov SI. Tumor promotion via injury- and death-induced inflammation. Immunity. 2011;35:467–77.
Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–99.
Widera D, Mikenberg I, Elvers M, Kaltschmidt C, Kaltschmidt B. Tumor necrosis factor alpha triggers proliferation of adult neural stem cells via IKK/NF-kappaB signaling. BMC Neurosci. 2006;20:7–64.
Velnar T, Bailey T, Smrkolj V. The wound healing process: an overview of the cellular and molecular mechanisms. J Int Med Res. 2009;37:1528–42.
Van Linthout S, Miteva K, Tschöpe C. Crosstalk between fibroblasts and inflammatory cells. Cardiovasc Res. 2014;102:258–69.
Beurel E, Michalek SM, Jope RS. Innate and adaptive immune responses regulated by glycogen synthase kinase-3 (GSK3). Trends Immunol. 2010;31:24–31.
Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol. 2005;6:777–84.
Nava P, Koch S, Laukoetter MG, Lee WY, Kolegraff K, Capaldo CT, et al. Interferon-gamma regulates intestinal epithelial homeostasis through converging beta-catenin signaling pathways. Immunity. 2010;32:392–402.
Dees C, Distler JH. Canonical Wnt signalling as a key regulator of fibrogenesis - implications for targeted therapies? Exp Dermatol. 2013;22:710–3.
Guo Y, Xiao L, Sun L, Liu F. Wnt/beta-catenin signaling: a promising new target for fibrosis diseases. Physiol Res. 2012;61:337–46.
Lam AP, Gottardi CJ. β-catenin signaling: a novel mediator of fibrosis and potential therapeutic target. Curr Opin Rheumatol. 2011;23:562–7.
Cheon SS, Cheah AY, Turley S, Nadesan P, Poon R, Clevers H, et al. beta-Catenin stabilization dysregulates mesenchymal cell proliferation, motility, and invasiveness and causes aggressive fibromatosis and hyperplastic cutaneous wounds. Proc Natl Acad Sci U S A. 2002;99:6973–8.
Verjee LS, Verhoekx JS, Chan JK, Krausgruber T, Nicolaidou V, Izadi D, et al. Unraveling the signalling pathways promoting fibrosis in Dupuytren's disease reveals TNF as a therapeutic target. Proc Natl Acad Sci U S A. 2013;110:E928–37.
Zhou B, Liu Y, Kahn M, Ann DK, Han A, Wang H, et al. Interactions between β-catenin and transforming growth factor-β signaling pathways mediate epithelial-mesenchymal transition and are dependent on the transcriptional co-activator cAMP-response element-binding protein (CREB)-binding protein (CBP). J Biol Chem. 2012;287:7026–38.
Escobar C, Munker R, Thomas JO, Li BD, Burton GV. Update on desmoid tumors. Ann Oncol. 2012;23:562–9.
Cohen S, Ad-El D, Benjaminov O, Gutman H. Post-traumatic soft tissue tumors: case report and review of the literature a propos a post-traumatic paraspinaldesmoid tumor. World J Surg Oncol. 2008;6:28.
Tejpar S, Nollet F, Li C, Wunder JS, Michils G, Dal Cin P, et al. Predominance of beta-catenin mutations and beta-catenin dysregulation in sporadic aggressive fibromatosis (desmoid tumor). Oncogene. 1999;18:6615–20.
Lazar AJ, Tuvin D, Hajibashi S, Habeeb S, Bolshakov S, Mayordomo-Aranda E, et al. Specific mutations in the beta-catenin gene (CTNNB1) correlate with local recurrence in sporadic desmoid tumors. Am J Pathol. 2008;173:1518–27.
Meneghello C, Ousghir B, Rastrelli M, Anesi L, Sommariva A, Montesco MC, et al. Nuclear GSK-3β segregation in desmoid-type fibromatosis. Histopathology. 2013;62:1098–108.
Caspi M, Zilberberg A, Eldar-Finkelman H, Rosin-Arbesfeld R. Nuclear GSK-3beta inhibits the canonical Wnt signalling pathway in a beta-catenin phosphorylation-independent manner. Oncogene. 2008;27:3546–55.
Yook JI, Li XY, Ota I, Hu C, Kim HS, Kim NH, et al. A Wnt-Axin2-GSK3beta cascade regulates Snail1 activity in breast cancer cells. Nat Cell Biol. 2006;8:1398–406.
Locci P, Bellocchio S, Lilli C, Marinucci L, Cagini L, Baroni T, et al. Synthesis and secretion of transforming growth factor-b1 by human desmoid fibroblast cell line and its modulation by toremifene. J Interferon Cytokine Res. 2001;21:961–70.
Ferenc T, Stalińska L, Turant M, Sygut J, Tosik D, Dziki A, et al. Analysis of TGF-beta protein expression in aggressive fibromatosis (desmoid tumor). Pol J Pathol. 2006;57:77–81.
Amini Nik S, Ebrahim RP, Van Dam K, Cassiman JJ, Tejpar S. TGFb modulates β-catenin stability and signaling in mesenchymal proliferations. Exp Cell Res. 2007;313:2887–95.
Mignemi NA, Itani DM, Fasig JH, Keedy VL, Hande KR, Whited BW, et al. Signal transduction pathway analysis in desmoid-type fibromatosis: transforming growth factor-β, COX2 and sex steroid receptors. Cancer Sci. 2012;103:2173–80.
Khurana JS, Ogino S, Shen T, Parekh H, Scherbel U, DeLong W, et al. Bone morphogenetic proteins are expressed by both bone-forming and non-bone-forming lesions. Arch Pathol Lab Med. 2004;128:1267–9.
Colombo C, Creighton CJ, Ghadimi MP, Bolshakov S, Warneke CL, Zhang Y, et al. Increased midkine expression correlates with desmoid tumour recurrence:a potential biomarker and therapeutic target. J Pathol. 2011;225:574–82.
Liegl B, Leithner A, Bauernhofer T, Windhager R, Guelly C, Regauer S, et al. Immunohistochemical and mutational analysis of PDGF and PDGFR in desmoid tumours: is there a role for tyrosine kinase inhibitors in c-kit-negative desmoid tumours? Histopathology. 2006;49:576–81.
Ishizuka M, Hatori M, Dohi O, Suzuki T, Miki Y, Tazawa C, et al. Expression profiles of sex steroid receptors in desmoid tumors. Tohoku J Exp Med. 2006;210:189–98.
Leithner A, Gapp M, Radl R, Pascher A, Krippl P, Leithner K, et al. Immunohistochemical analysis of desmoid tumours. J Clin Pathol. 2005;58:1152–6.
Beyer C, Reichert H, Akan H, Mallano T, Schramm A, Dees C, et al. Blockade of canonical Wnt signalling ameliorates experimental dermal fibrosis. Ann Rheum Dis. 2013;72:1255–8.
Ren S, Johnson BG, Kida Y, Ip C, Davidson KC, Lin SL, et al. LRP-6 is a coreceptor for multiple fibrogenic signalling pathways in pericytes and myofibroblasts that are inhibited by DKK-1. Proc Natl Acad Sci U S A. 2013;110:1440–5.
Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA, et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461:614–20.
Wang C, Zhu H, Sun Z, Xiang Z, Ge Y, Ni C, et al. Inhibition of Wnt/β-catenin signaling promotes epithelial differentiation of mesenchymal stem cells and repairs bleomycin-induced lung injury. Am J Physiol Cell Physiol. 2014;307:C234–44.
Lau Chan E, Callow M, Waaler J, Boggs J, Blake RA, Magnuson S, et al. A novel tankyrase small-molecule inhibitor suppresses APC mutation-driven colorectal tumor growth. Cancer Res. 2013;73:3132–44.
Abu-Baker A, Laganiere J, Gaudet R, Rochefort D, Brais B, Neri C, et al. Lithium chloride attenuates cell death in oculopharyngeal muscular dystrophy by perturbing Wnt/β-catenin pathway. Cell Death Dis. 2013;4:e821.
Hu X, Paik PK, Chen J, Yarilina A, Kockeritz L, Lu TT, et al. IFN-γ suppresses IL-10 production and synergizes with TLR2 by regulating GSK3 and CREB/AP-1 proteins. Immunity. 2006;24:563–74.
Whittle BJ, Varga C, Posa A, Molnar A, Collin M, Thiemermann C. Reduction of experimental colitis in the rat by inhibitors of glycogen synthase kinase-3β. Br J Pharmacol. 2006;147:575–82.
de Bree E, Zoras O, Hunt JL, Takes RP, Suárez C, Mendenhall WM, et al. Desmoid tumors of the head and neck: A therapeutic challenge. Head Neck. 2014;36:1517–26.
Walczak BE, Rose PS. Desmoid: the role of local therapy in an era of systemic options. Curr Treat Options Oncol. 2013;3:465–73.
Bonvalot S, Ternès N, Fiore M, Bitsakou G, Colombo C, Honoré C, et al. Spontaneous regression of primary abdominal wall desmoid tumors: more common than previously thought. Ann Surg Oncol. 2013;20:4096–102.
Fiore M, Rimareix F, Mariani L, Domont J, Collini P, Le Péchoux C, et al. Desmoid-type fibromatosis: a front-line conservative approach to select patients for surgical treatment. Ann Surg Oncol. 2009;16:2587–93.
Lev D, Kotilingam D, Wei C, Ballo MT, Zagars GK, Pisters PW, et al. Optimizing treatment of desmoid tumors. J Clin Oncol. 2007;25:1785–91.
Ballo MT, Zagars GK, Pollack A, Pisters PW, Pollack RA. Desmoid tumor: prognostic factors and outcome after surgery, radiation therapy, or combined surgery and radiation therapy. J Clin Oncol. 1999;17:158–67.
Melis M, Zager JS, Sondak VK. Multimodality management of desmoid tumors: how important is a negative surgical margin? J Surg Oncol. 2008;98:594–602.
Ballo MT, Zagars GK, Pollock RE, Benjamin RS, Feig BW, Cormier JN, et al. Retroperitoneal soft tissue sarcoma: an analysis of radiation and surgical treatment. Int J Radiat Oncol Biol Phys. 2007;67:158–63.
Janinis J, Patriki M, Vini L, Aravantinos G, Whelan JS. The pharmacological treatment of aggressive fibromatosis: a systematic review. Ann Oncol. 2003;14:181–90.
Leithner A, Schnack B, Katterschafka T, Wiltschke C, Amann G, Windhager R, et al. Treatment of extra-abdominal desmoid tumors with interferon-alpha with or without tretinoin. J Surg Oncol. 2000;73:21–5.
Poritz LS, Blackstein M, Berk T, Gallinger S, McLeod RS, Cohen Z. Extended follow-up of patients treated with cytotoxic chemotherapy for intra-abdominal desmoid tumors. Dis Colon Rectum. 2001;44:1268–73.
Penel N, Le Cesne A, Bui BN, Perol D, Brain EG, Ray-Coquard I, et al. Imatinib for progressive and recurrent aggressive fibromatosis (desmoid tumors): an FNCLCC/French Sarcoma Group phase II trial with a long-term follow-up. Ann Oncol. 2011;22:452–7.
Gounder MM, Lefkowitz RA, Keohan ML, D'Adamo DR, Hameed M, Antonescu CR, et al. Activity of Sorafenib against desmoid tumor/deep fibromatosis. Clin Cancer Res. 2011;17:4082–90.
Mace J, Sybil Biermann J, Sondak V, McGinn C, Hayes C, Thomas D, et al. Response of extraabdominal desmoid tumors to therapy with imatinib mesylate. Cancer. 2002;95:2373–9.
Stabellini G, Balducci C, Lilli C, Marinucci L, Becchetti E, Carinci F, et al. Toremifene decreases type I, type II and increases type III receptors in desmoid and fibroma and inhibits TGFbeta1 binding in desmoid fibroblasts. Biomed Pharmacother. 2008;62:436–42.
Ghanbari-Azarnier R, Sato S, Wei Q, Al-Jazrawe M, Alman BA. Targeting stem cell behavior in desmoid tumors (aggressive fibromatosis) by inhibiting hedgehog signaling. Neoplasia. 2013;15:712–9.
Issakov J, Merimsky O, Gutman M, Kollender Y, Lev-Chelouche D, Abu-Abid S, et al. Hyperthermic isolated limb perfusion with tumor necrosis factor-alpha and melphalan in advanced soft-tissue sarcomas: histopathological considerations. Ann Surg Oncol. 2000;7:155–9.
Drouet A, Le Moigne F, Have L, Blondet R, Jacquin O, Chauvin F. Common peroneal nerve palsy following TNF-based isolated limb perfusion for irresectable extremity desmoid tumor. Orthop Traumatol Surg Res. 2009;95:639–44.
Bonvalot S, Rimareix F, Causeret S, Le Péchoux C, Boulet B, Terrier P, et al. Hyperthermic isolated limb perfusion in locally advanced soft tissue sarcoma and progressive desmoid-type fibromatosis with TNF 1 mg and melphalan (T1-M HILP) is safe and efficient. Ann Surg Oncol. 2009;16:3350–7.
Acknowledgements
We thank the “Mauro Baschirotto” Institute for Rare Diseases for its continuous support of our research. Our deep thanks go to the Desmon association for their support and collaboration.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors do not have any conflicts of interests.
Authors’ contributions
MVE: Wnt pathway actors, Wnt pathway in association to diseases. MR: therapeutic approaches for desmoid type fibromatosis. CRR: therapeutic approaches for desmoid type fibromatosis. UH: genetic aspects of Wnt pathway diseases. DS: Wnt pathway in inflammation, Wnt pathway in association to diseases. All authors read and approved the final manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Enzo, M.V., Rastrelli, M., Rossi, C.R. et al. The Wnt/β-catenin pathway in human fibrotic-like diseases and its eligibility as a therapeutic target. Mol and Cell Ther 3, 1 (2015). https://doi.org/10.1186/s40591-015-0038-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40591-015-0038-2